
水诱导磷化镍腐蚀及腐蚀抑制
2022-07-12 07:40:08吕功煊张旭强马建泰
无机化学学报 2022年7期
杨 博 吕功煊 张旭强 马建泰
(1中国科学院兰州化学物理研究所,羰基合成与选择氧化国家重点实验室,兰州 730000)(2中国科学院大学,北京 100049)(3兰州大学化学化工学院,兰州 730000)
0 引 言
过渡金属磷化物作为一种高活性、可替代贵金属的材料在催化、电催化和光(电)催化等领域作为共催化剂有着广泛应用[1-4]。20世纪90年代,研究人员发现无定型的Ni-Px和Ni-Co-P合金具有高的电催化水分解制氢和氧活性[5-6]。理论和实验研究均表明,过渡金属磷化物(如Ni2P、FeP、Ni5P4和CoP等)在电催化析氢反应(HER)过程中有低的过电势和高的循环伏安稳定性[7-11]。同时研究也表明,过渡金属磷化物在阳极极化的条件下,发生表面部分氧化重构后会形成具有析氧反应(OER)活性的催化剂[12-13]。因此过渡金属磷化物也被用作水分解的双功能催化剂,实现高效水分解产氢和产氧[14-15]。由于过渡金属磷化物具有优异的催化特性,也常被用作光催化助催化剂,降低水分解过电位,提高半导体表面载流子的分离效率,如 FeP/TiO2、Co2P/CdS、Ni2P/CdS、Co@Co2P/RGO等光催化剂均表现出高的产氢效率和稳定性[16-19]。
但也有研究指出,过渡金属磷化物在催化反应过程中不稳定,这可能成为制约其应用的一个重要因素。Lewis等研究表明CoP在电催化HER反应中表面可能存在无定型的Co和P物种[20-21]。Bard指出CoP在HER过程中存在着表面氧化和酸腐蚀过程[22]。Kucernak研究表明磷化镍在酸性溶液中的腐蚀程度与其中P含量相关,高磷含量NixP在酸中的腐蚀程度较低[23]。同样也有报道指出,过渡金属磷化物作为助催化剂制备的复合光催化剂催化活性不稳定。如Cu3P/CNT催化剂在曙红-三乙醇胺体系中经过4次循环反应后,其催化活性下降了20%[24];g-C3N4-CoP-K2HPO4体系在经过5次光催化循环后其催化活性损失25%[25]。
目前为止,过渡金属磷化物作为电催化或光催化助剂长周期活性出现下降的原因仍不是完全清楚。因此,若能对过渡金属磷化物在水溶液中的腐蚀机理进一步深入理解,并能够提出一种保护策略,对其作为电催化电极和光催化助剂的研究来说具有重要意义。我们以NixP作为过渡金属磷化物的典型代表,研究了过渡金属磷化物在水介质中的腐蚀规律及抑制策略。采用低温磷化法通过调节前驱体NaH2PO2和NiCl2的物质的量比值(nP/nNi,文中如无特别说明,均指初始nP/nNi)合成了系列NixP,并测试了其在暗态水介质中的稳定性。结果表明NixP在水介质中不稳定,其与水反应生成H2,同时自身被氧化产生PO43-和Ni2+,发生化学腐蚀。不同nP/nNi合成的NixP因在晶型、形貌和组成方面的差异而表现出不同的腐蚀行为。在NixP表面包覆惰性保护层NiO、ZnO和TiO2,一定程度上可抑制NixP在水中的腐蚀。
1 实验部分
1.1 材料的制备
1.1.1 NixP的制备
采用低温固态磷化法合成不同nP/nNi的NixP。将 NiCl2·6H2O(6 mmol)和 NaH2PO2·H2O(36、24、12、6或3 mmol)充分混合研磨,得到绿色胶状混合物。为了去除混合物中的结晶水,将其转移至鼓风干燥箱中100℃处理3 h。再次研磨混合物后,将粉末样品转移至瓷舟并置于石英管式炉中,在高纯Ar气保护下300℃热处理2 h。反应系统冷却至室温,得到黑色固体粉末,经过洗涤干燥后得到NixP样品。
1.1.2 NiO@NixP、ZnO@NixP和TiO2@NixP的制备
NiO@NixP的制备采用空气氛围高温快速退火法。将200 mg NixP(nP/nNi=2)置于坩埚中并快速置于预升温至450℃的马弗炉中,快速退火1、2、5、10 min制备NixP(nP/nNi=2)表面氧化的NiO@NixP材料。
以0.1 mol·L-1醋酸锌的乙醇溶液作为锌源,通过调节加入醋酸锌溶液的体积(25、74、123、246 μL),制备不同厚度ZnO包裹的ZnO@NixP复合材料。将200 mg NixP(nP/nNi=2)超声分散于无水乙醇中,滴加一定体积的0.1 mol·L-1醋酸锌的乙醇溶液,并将混合溶液转移至50℃的鼓风干燥箱中彻底干燥。随后将干燥的混合物置于Ar气保护的石英管式炉中300℃恒温处理60 min,并自然冷却至室温。根据ZnO含量将样品标记为m-ZnO@NixP,其中m代表ZnO在复合材料中的质量分数(m=0.1%、0.3%、0.5%、1.0%)。
基于相同的制备方法,利用钛酸四丁酯作为钛源,合成了不同厚度TiO2包裹NixP(nP/nNi=2)的复合材料,且所得样品标记为n-TiO2@NixP,其中n代表TiO2在复合材料中的质量分数(n=0.1%、0.3%、0.5%、1.0%)。NixP和ZnO@NixP、TiO2@NixP的制备流程如图1所示。
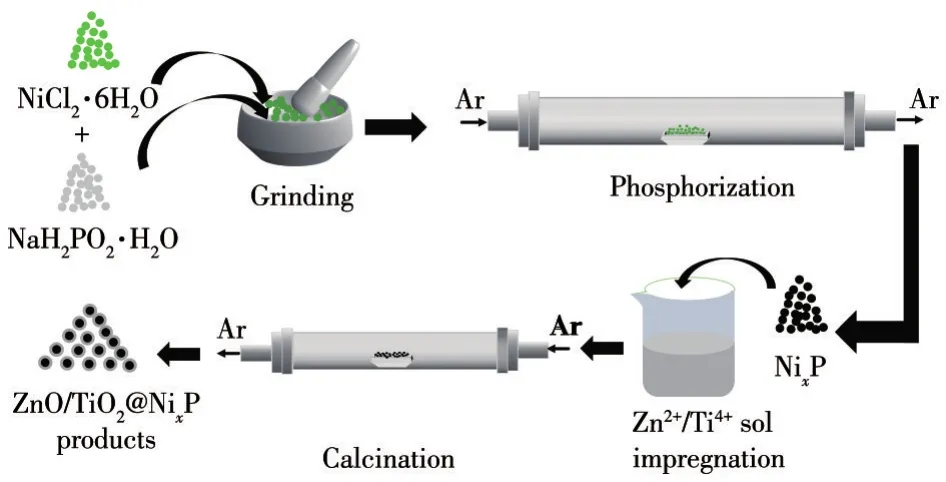
图1 NixP、ZnO@NixP、TiO2@NixP的制备Fig.1 Synthesis of NixP,ZnO@NixP,and TiO2@NixP
1.2 水介质中NixP稳定性测试
采用一个顶端硅胶密封的石英瓶作为反应器,检测NixP在水介质中稳定性。将50 mg NixP置于石英反应器中,加入150 mL去离子水并超声分散10 min。利用高纯Ar置换30 min以除去体系中残余的空气。在暗态条件下每隔相同时间从反应器中抽取0.5 mL的气体,利用Agilent气相色谱测定其气相组成及含量。为了证实NixP在水介质中的腐蚀反应与光照条件无关,在相同的反应条件下将石英反应瓶置于一个磁力搅拌器上搅拌,采用300 W Xe灯(配备420 nm截止滤光片)作为光源照射反应体系。每隔相同时间从反应器中抽取0.5 mL的气体,利用Agilent气相色谱测定其气相组成及含量。
1.3 反应液中PO4 3-浓度的测试
溶液中PO43-浓度采用钼酸铵分光光度法进行测定[26]。分别移取 0.0、0.5、1.0、3.0、5.0、7.0、9.0、11.0、13.0 mL KH2PO4(含磷2 μg·mL-1)标准溶液于具塞试管中,加入1.0 mL抗坏血酸(100 mg·mL-1)和2.0 mL钼酸铵-酒石酸锑钾混合溶液(钼酸铵浓度26 mg·mL-1,酒石酸锑钾浓度 0.7 mg·mL-1)。分别用去离子水稀释至20 mL并在室温下静置20 min,随后利用紫外可见分光光度计在700 nm波长下分别测定各溶液的吸光度,获得PO43-的标准曲线。反应溶液中PO43-浓度测定的具体操作如下:取离心处理的反应溶液1 mL,随后加入等量的抗坏血酸和钼酸铵-酒石酸锑钾混合溶液并稀释至20 mL,摇晃均匀后静置20 min测其吸光度值。根据标准曲线,计算反应溶液中PO43-浓度。
1.4 材料的表征
采用日本Rigaku公司生产的X射线衍射仪(XRD,Smartlab-SE X-ray diffractometer)测定合成材料的晶体特性,工作电压40 kV,工作电流40 mA,辐射源为CuKα靶,波长为0.154 nm,扫描范围2θ=10°~80°。采用FEI公司生产的透射电子显微镜(TEM,Tecnai-G2-TF20)测试材料的形貌,电子束工作电压为200 kV。样品超声分散于无水乙醇中,将分散悬浮液滴于Cu网上,自然缓慢干燥后测试。采用ThermoFisher Scientific公司生产的X射线光电子能谱(XPS,ESCALAB 250 Xi)分析材料表面的元素组成及价态,辐射源为单色AlKα(hν=1 486.6 eV),元素结合能采用C1s(284.8 eV)进行校正。采用PANalytical生产的X射线荧光光谱仪(XRF,Magix PW2403)分析合成材料中体相元素的含量。采用上海仪器分析厂生产的751G分光光度计测定反应溶液中的PO3-4浓度,光源为钨灯。采用Agilent公司生产的6820气相色谱检测反应体系中的气体种类及含量。采用Agilent公司制造的电感耦合等离子体发射光谱仪(ICP-OES,Agilent 725-ES)测量溶液中总 Ni、P元素的浓度。
2 结果与讨论
2.1 水诱导NixP腐蚀
图2a为不同nP/nNi合成的NixP催化体系在暗态下的析氢情况。测试结果表明,所有反应体系均检测到H2,且随着反应时间的延长H2含量逐渐增多。值得注意的是,不同nP/nNi合成的NixP在相同反应条件下的析氢速率并不相同,nP/nNi=0.5、1、2、4、6时合成的NixP催化体系在水中反应3 h的平均析氢速率(rH2)分别为 0.58、0.92、1.14、4.21、11.37 μmol·h-1(图2b)。因此可以确定NixP在水介质中不稳定,极易与水发生化学反应产生氢气,且析氢速率正比于nP/nNi。此外,nP/nNi=2合成的NixP在暗态水环境中的循环析氢活性如图2c所示,NixP在多次循环过程中析氢活性基本保持稳定,证明其与水发生化学腐蚀,而非表面吸附物的溶解。
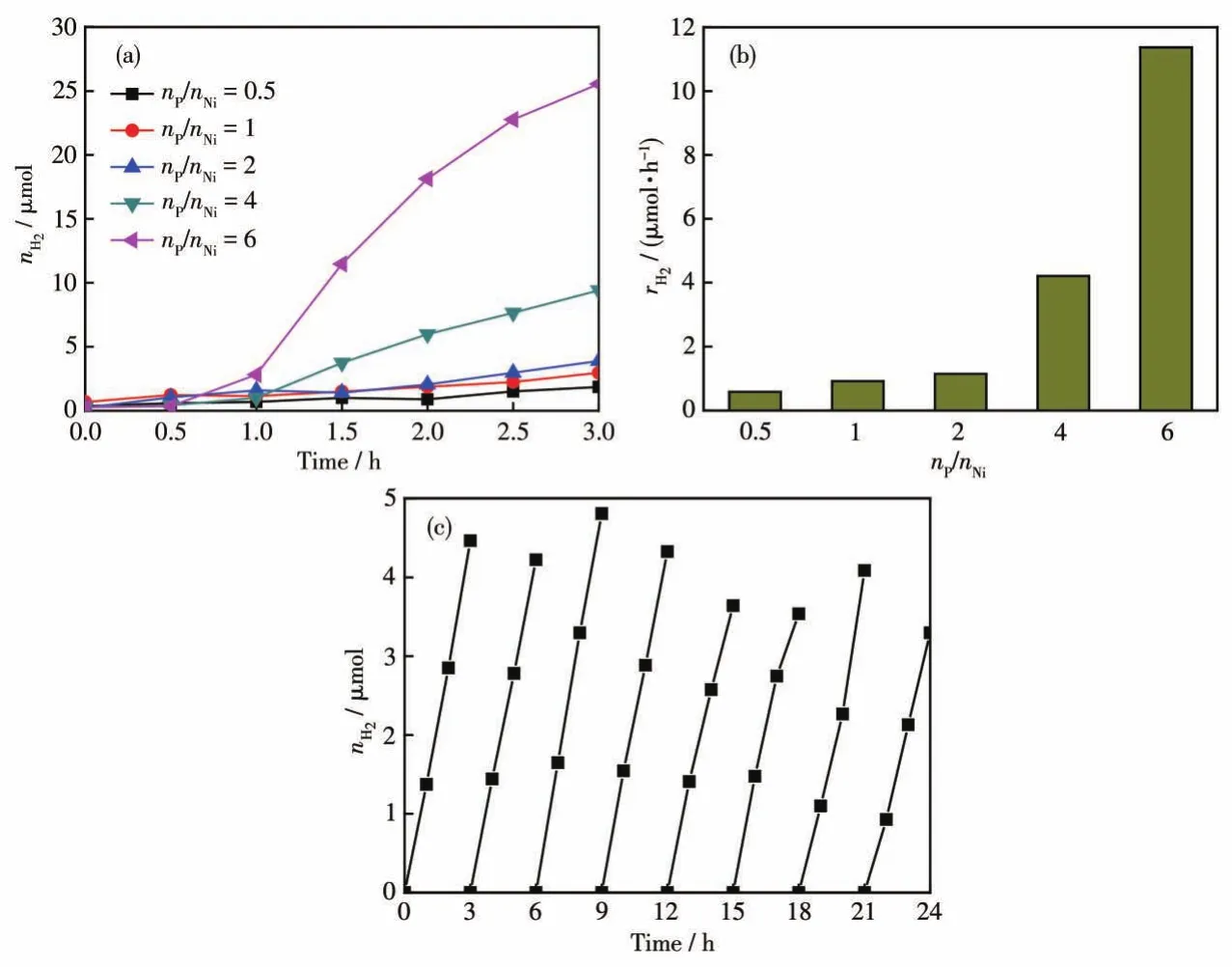
图2 (a)不同nP/nNi时合成的NixP在水中暗态条件下的析氢活性;(b)相应体系在水中的rH2;(c)nP/nNi=2合成的NixP在水中暗态条件下的析氢循环稳定性Fig.2 (a)Hydrogen evolution activity of NixP synthesized with different nP/nNiin the dark;(b)rH2of corresponding NixP in the dark;(c)Cycling stability of hydrogen evolution activity for NixP(nP/nNi=2)in the dark
在检测体系气相组成的同时,采用分光光度法和ICP-OES法检测了相应体系在暗态反应过程中溶液中PO43-和Ni2+浓度的变化。分光光度法测试溶液中PO43-浓度变化的结果如图3a所示,所有反应体系中均检测到PO43-,且随着反应时间的延长,溶液中PO43-的浓度逐渐增加。nP/nNi=0.5、1、2、4、6时合成的NixP与H2O反应3 h后,溶液中PO43-的浓度分别为 21.54、16.50、4.50、7.32、7.91 mg·L-1。显然随着nP/nNi的升高,反应溶液中的PO43-浓度先降低后升高,并且nP/nNi=2时合成的NixP在水中检测到的PO43-浓度最低,意味着在此条件下制备的NixP结构较为稳定。这一结论与ICP-OES测试溶液中P和Ni物种的变化一致(图3b)。为了进一步研究NixP在水介质中的稳定性,在Xe灯光照条件下,测试了NixP的析氢活性和溶液中PO43-浓度。nP/nNi=0.5、1、2、4、6时合成的NixP在光照下的平均产氢速率(rH2)分别为0.46、0.62、0.71、2.17、9.44 μmol·h-1(图3c);反应后溶液中的PO43-浓度分别为15.53、13.84、5.64、6.64、5.56 mg·L-1(图3d)。测试结果表明:不同nP/nNi合成的NixP在光照和暗态条件下的反应规律一致,光照并未加快NixP在H2O中的腐蚀。因此,基于上述的析氢活性测试和光谱表征,可以证实NixP在水环境中极易发生化学腐蚀,且这种腐蚀速率取决于NixP的组成比例或晶体结构,腐蚀反应在产生H2的同时,NixP中的磷物种被氧化生成PO43-。
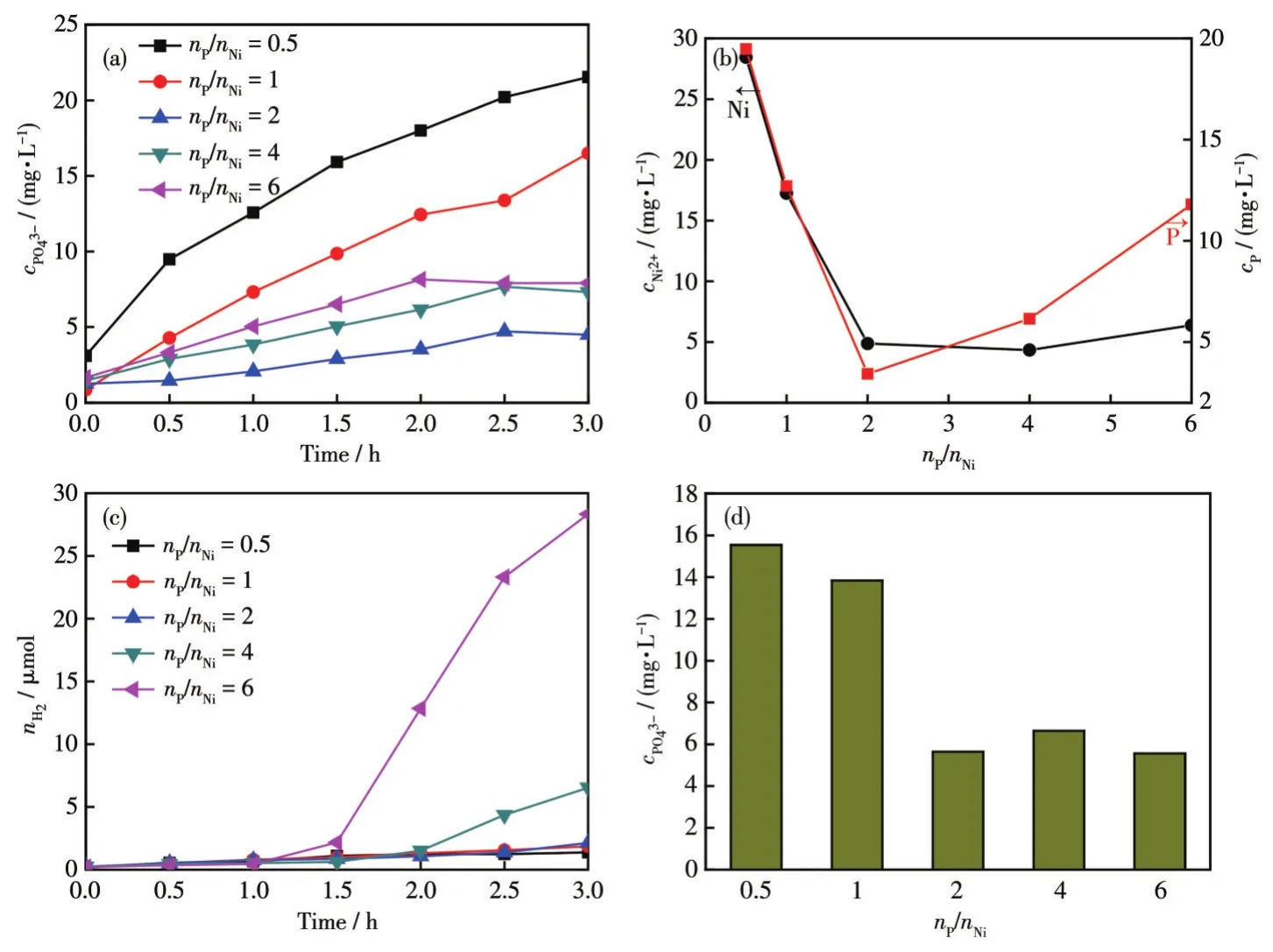
图3 不同nP/nNi时合成的NixP:(a)分光光度法检测暗态反应条件下溶液中PO43-浓度随时间变化的曲线;(b)ICP-OES检测暗态反应3 h后溶液中Ni和P物种的浓度;(c)光照条件下NixP的析氢活性;(d)分光光度法检测光照反应3 h后溶液中PO43-浓度Fig.3 NixP synthesized with different nP/nNi:(a)concentration of phosphate ions versus time in NixP reaction system measured by spectrophotometry in the dark;(b)concentration of Ni and P measured by ICP-OES in NixP reaction system after 3 h reaction in the dark;(c)H2evolution activity of NixP under light irradiation;(d)spectrophotometric detection of PO43-concentration in solution after 3 h light irradiation reaction
以NixP(nP/nNi=2)为例,进一步研究了其在水中暗态循环反应前后表面形貌、晶体结构及元素状态的变化规律。从TEM和HRTEM图对比不难发现,NixP纳米颗粒经历循环反应之后表面变得无序,这降低了颗粒表面结晶度(图4a~4d)。样品的高角环形暗场扫描透射电子显微镜像(HAADF-STEM)及元素分布图(图4e~4l)表明,反应前NixP表面Ni和P元素分布均匀,但O元素含量较低。经历水循环反应后NixP表面的O含量明显增加,意味着NixP与水发生化学腐蚀,表面形成少量无定型的氢氧化物覆盖在样品表面。循环反应前后的XRD图中仅包含NixP的衍射峰,表明反应后NixP的晶体结构并未改变,但少量无定型氢氧化物的包裹导致NixP衍射峰强度降低(图4m)。基于上述的实验结果可以断定NixP在水介质中不稳定,其自发与H2O反应生成H2和PO43-的同时,表面生成少量无定型氢氧化物。可能的反应如下:
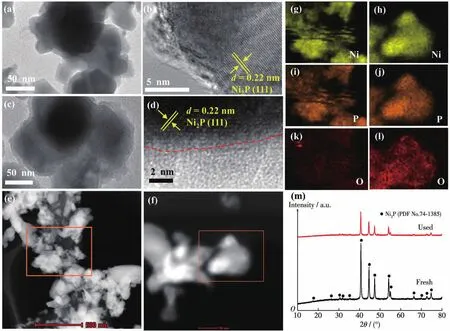
图4 NixP(nP/nNi=2)的TEM图 (a)、HRTEM图 (b)、HAADF-STEM图 (e)和Ni、P、O元素分布图 (g、i、k);NixP(nP/nNi=2)循环实验后的TEM图 (c)、HRTEM图 (d)、HAADF-STEM图 (f)和Ni、P、O元素分布图 (h、j、l);NixP(nP/nNi=2)循环反应前后的XRD图(m)Fig.4 TEM image(a),HRTEM image(b),HAADF-STEM image(e),and Ni,P,O element mappings(g,i,k)of NixP(nP/nNi=2);TEM image(c),HRTEM image(d),HAADF-STEM image(f),and Ni,P,O element mappings(h,j,l)of NixP(nP/nNi=2)after cycling reaction;XRD patterns of NixP(nP/nNi=2)after cycling reaction(m)
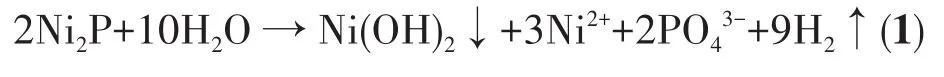
2.2 不同nP/nNi合成的NixP的结构表征
从不同nP/nNi合成的NixP在水中腐蚀的析氢量与离子浓度分析中不难发现,随着nP/nNi的增加,析氢速率逐渐增大,而离子浓度的变化趋势却是先降低后升高,两者的变化规律存在很大差异。这意味着合成的NixP由于组成和结构等特性的不同,导致其在水中的腐蚀速率不同。我们以NaH2PO2·H2O和 NiCl2·6H2O为前驱体,通过调控nP/nNi,在较低反应温度下成功制备了组分不同的NixP,其可能的反应可用式2~6表示。NiCl2与NaH2PO2发生置换反应得到Ni(H2PO2)2,其进一步分解生成PH3和NiHPO4,随后 NiHPO4、PH3、NiCl2再次反应得到NixP化合物。从反应式中不难发现,Ni物种的价态保持不变,因此可以推断在NixP的化学腐蚀过程中,Ni物种对于腐蚀反应中的产氢没有贡献,腐蚀析氢仅与P物种相关。
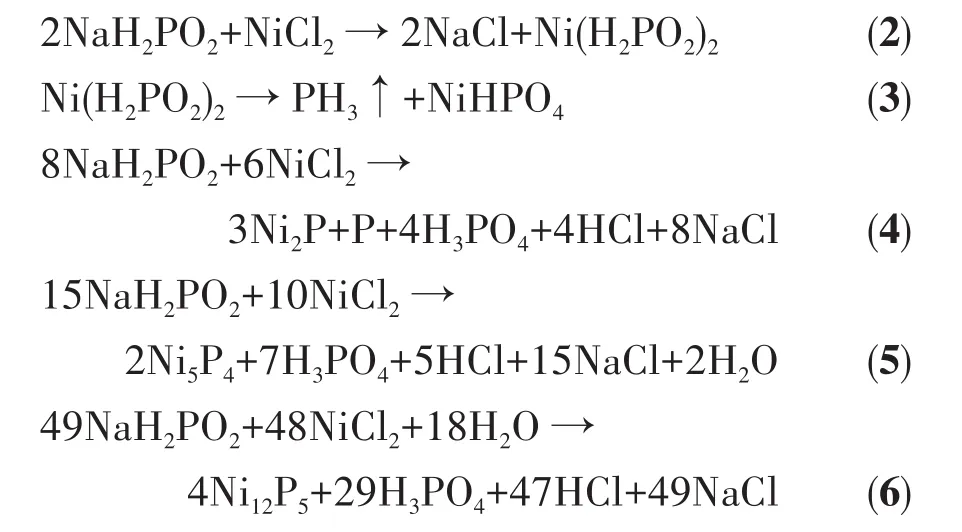
为了揭示不同nP/nNi合成的NixP在水介质中腐蚀性能的差异,首先利用XRD对所有样品的晶体结构进行了表征,其结果如图5所示。当nP/nNi=6和4时,样品包含2种不同物质的衍射峰。其中17.5°、30.5°、31.7°、35.3°、40.7°、44.6°、47.3°、54.2°、54.9°、66.4°、72.7°、74.8°处的峰为六方相Ni2P(PDF No.74-1385)[27],15.0°、16.2°、36.1°、52.9°处的峰为六方相Ni5P4(PDF No.18-0883)[28]。而当nP/nNi=2时,NixP 的XRD图显示为纯的六方相Ni2P。此外,当nP/nNi=1和0.5时制备的NixP为六方Ni2P和立方Ni12P5(PDF No.74-1381)混合物,其中2θ位于38.5°、41.7°、49.0°处的衍射峰对应立方相Ni12P5[29]。上述XRD结果表明,通过简单调控nP/nNi可获得结构不同的NixP。晶体结构及组分比例的差异或许是决定其在水介质中腐蚀速率的关键因素之一。
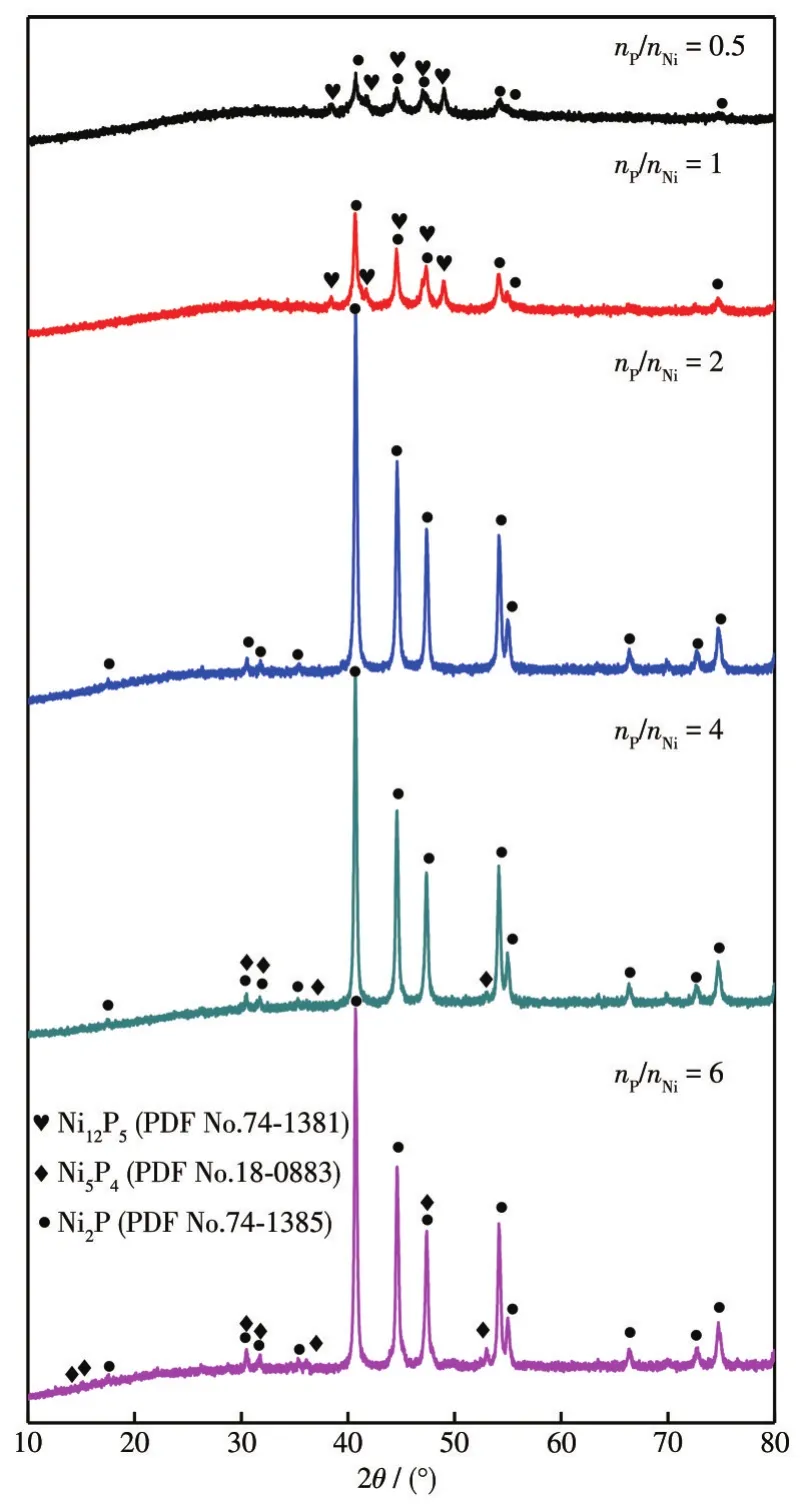
图5 不同nP/nNi时制备的NixP的XRD图Fig.5 XRD patterns of NixP synthesized with different nP/nNi
采用XPS进一步分析了NixP的表面元素组成和元素状态。如图6a所示,Ni2p3/2由位于约853.4、856.7、861.2 eV处的3个拟合峰构成,且分别归属于NixP 化合物中的 Niδ+、残留的 NiCl2和 Ni2p的卫星峰[30]。P2p可由位于约 129.5、130.5、133.4、134.4 eV处的4个峰组合而成,其中约129.5和130.5 eV处的峰归属于金属磷化物的P2p,结合能约133.4和134.4 eV处的拟合峰来源于残余磷酸盐(图6b)[31]。表1为Ni2p3/2和P2p拟合峰的面积占比,随着nP/nNi的增加,Niδ+的峰面积占比逐渐增大,同时位于约856.7 eV处的峰面积逐渐减小,表明通过调控nP/nNi可显著优化NixP表面Ni-P物种的占比。P2p高分辨XPS谱图的拟合结果表明,前驱体P的投入量与生成NixP表面Ni-P物种含量相关。由于nP/nNi不同,通过低温热处理法制备的NixP附着有未反应的Ni或P前驱体或中间产物,这些附着物在暗态水介质腐蚀反应过程中被溶解析出。这种物理溶解对ICP-OES和分光光度法的检测结果产生影响,但其不会改变材料在暗态水介质中腐蚀的析氢量。
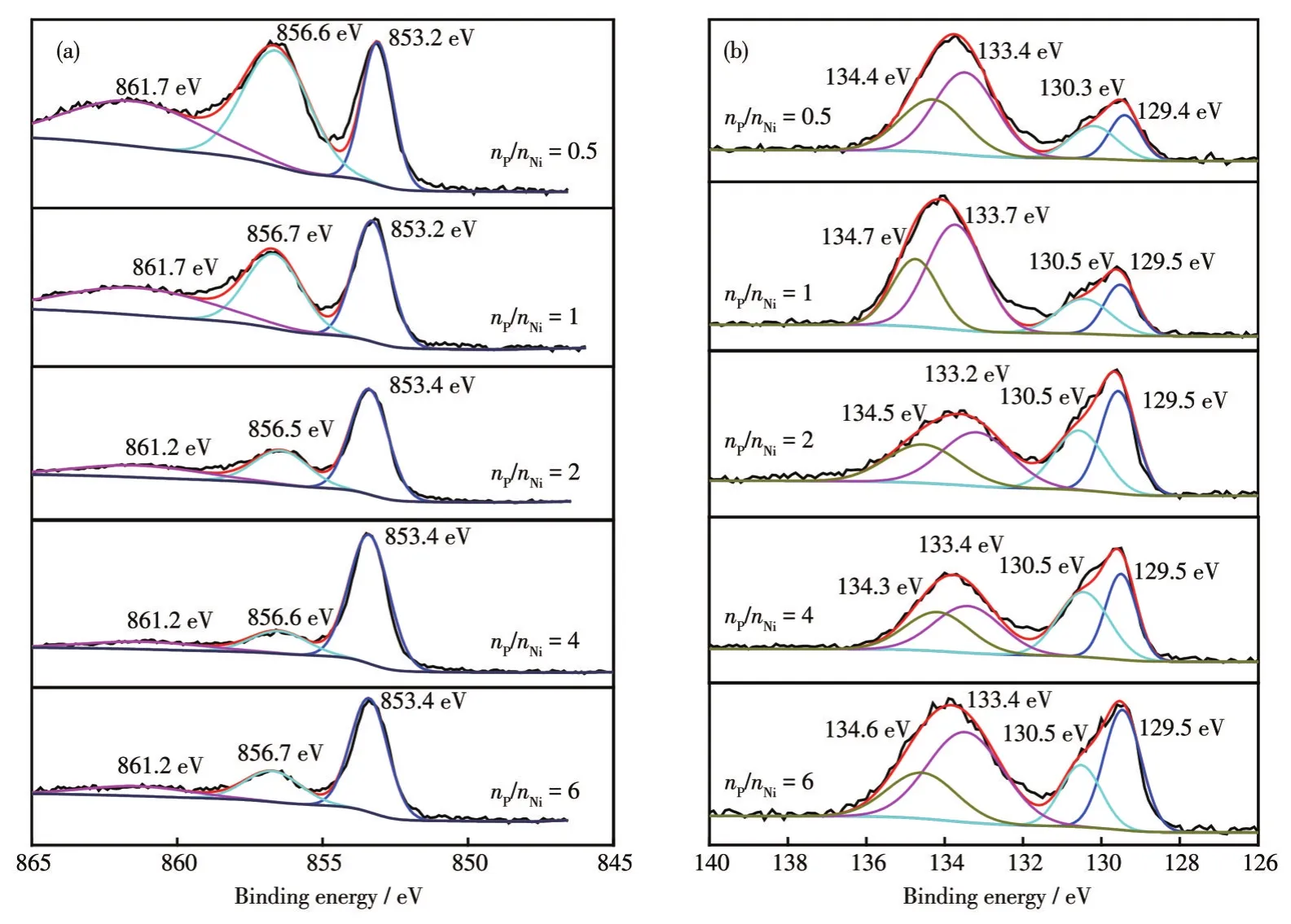
图6 不同nP/nNi时合成的NixP的(a)Ni2p3/2和(b)P2p XPS谱图Fig.6 XPS spectra of Ni2p3/2(a)and P2p(b)of NixP synthesized with different nP/nNi
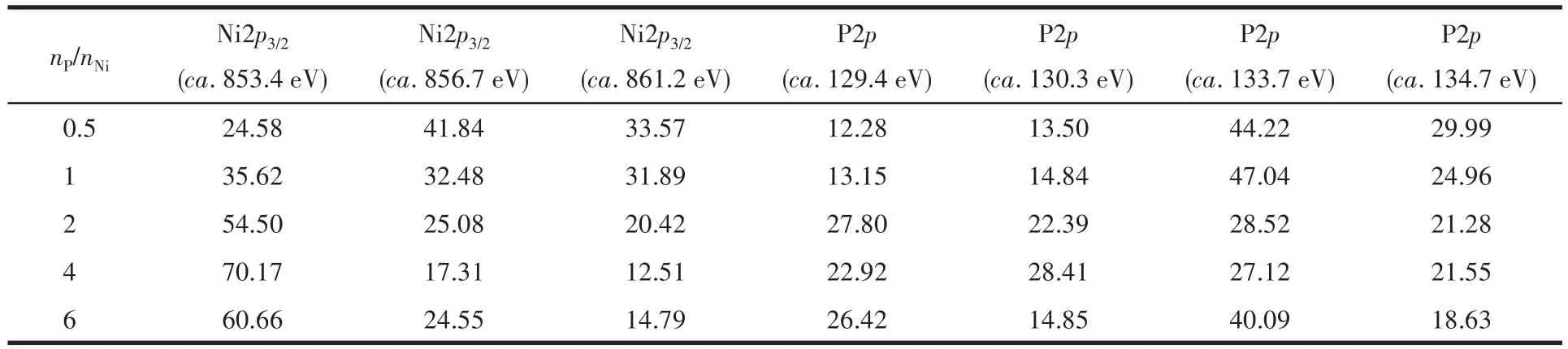
表1 不同nP/nNi时合成的NixP Ni2p3/2和P2p拟合峰面积占比(%)Table 1 Fraction(%)of Ni2p3/2and P2p fitting peak area of NixP synthesized with different nP/nNi
NixP的体相元素含量及比例可进一步通过XRF表征得到,结果如表2所示。当nP/nNi=2时,合成的NixP中nP/nNi=0.501,最接近Ni2P。而当升高nP/nNi时,XRF测得NixP中的nP/nNi>0.5,且随着nP/nNi增加,XRF测得NixP中的nP/nNi也升高。此外,当合成NixP的nP/nNi=0.5和1时,产物中nP/nNi也高于0.50。nP/nNi较低时意味着中间产物Ni3(PO4)2过量,而磷酸盐物种中nP/nNi比金属磷化物的高[32]。
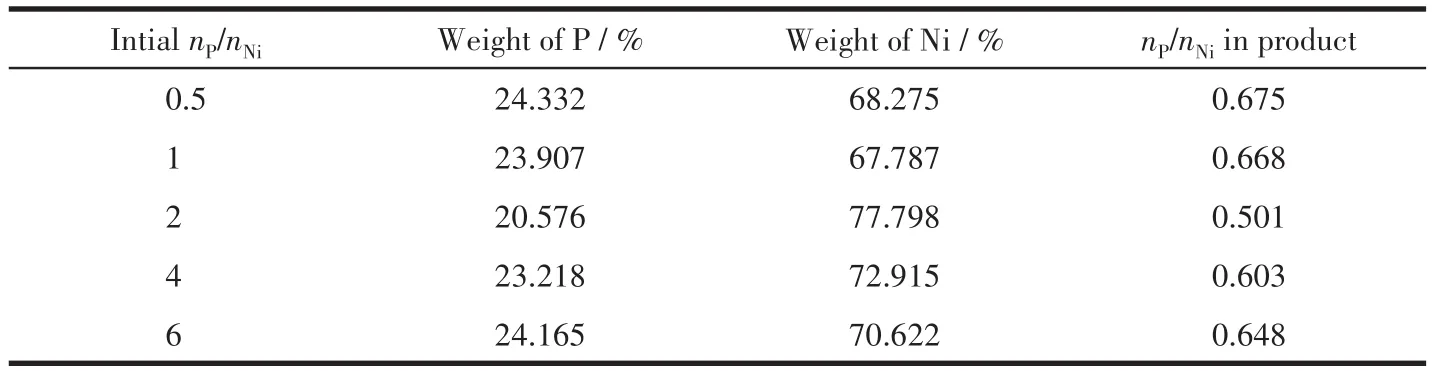
表2 利用XRF表征不同nP/nNi时合成的NixP中P和Ni的含量Table 2 XRF measurement of Ni and P content in NixP synthesized with different nP/nNi
图7是不同nP/nNi时合成NixP的TEM图。低nP/nNi时合成的NixP由较小的纳米颗粒组成(图7a和7c)。随着nP/nNi增加,NixP无规则的团聚物颗粒尺寸增大(图7e、7g、7i)。相应NixP的高倍TEM(HRTEM)图如图7b、7d、7f、7h、7j所示,nP/nNi=0.5时,NixP样品暴露的主要晶面晶格条纹间距为0.18和0.19 nm,分别归属于 Ni12P5的(312)晶面和 Ni2P 的(210)晶面[33-35]。NixP(nP/nNi=1)的HRTEM图暴露的晶格条纹间距为0.50和0.22 nm,其源自Ni2P的(100)和(111)晶面。而当nP/nNi=2、4和6时,样品暴露的晶格条纹间距为0.22 nm,归属于Ni2P的(111)晶面[36]。上述结果表明合成的NixP为多物相多晶面暴露的混合物,与XRD的表征结果一致。

图7 NixP的TEM和HRTEM图Fig.7 TEM and HRTEM images of NixP
2.3 包覆惰性保护层抑制NixP(nP/nNi=2)腐蚀
上述实验结果证明NixP在水介质中的稳定性不佳,会发生与水相关的化学腐蚀,其腐蚀速率与材料的晶体结构和元素比例密切相关。我们尝试在半导体材料表面生长一层薄的氧化物保护层以对其进行保护[37-44],构建高效稳定的NiO@NixP、ZnO@NixP、TiO2@NixP复合材料。在水参与的苛刻光(电)催化反应中,保护层应具有高的化学稳定性和耐光腐蚀能力,且易于透光和大规模制备。
将NixP(nP/nNi=2)在空气中直接煅烧,使其表面氧化形成一层薄的NiO保护层,获得稳定的NiO@NixP复合材料。图8a是NixP(nP/nNi=2)在空气中450℃退火不同时间制备NiO@NixP的Ni2p3/2精细XPS谱图。结合能位于约853.3 eV的Ni—P峰面积逐渐降低,同时位于约856.7 eV处的Ni—O峰面积占比逐渐增大,证明NixP(nP/nNi=2)表面被氧化并生成NiO薄膜,且随退火时间的延长保护层NiO厚度和覆盖率逐渐增加。相应NiO@NixP的XRD图如图8b所示,由于NixP(nP/nNi=2)被薄的NiO保护层包裹,致使磷化物的特征衍射峰强度降低。此外,退火时间为1、2、5、10 min制备的NiO@NixP在水介质暗态条件下的析氢速率分别为 2.45、1.12、1.25、0.80 μmol·h-1,相对于单一NixP(nP/nNi=2)的腐蚀析氢速率(2.33 μmol·h-1)降低(图8c)。通过调控退火时间,NixP(nP/nNi=2)表面的NiO保护层覆盖率增加,构筑的复合材料NiO@NixP与水发生反应析氢的几率降低,从而在一定程度上抑制了NixP(nP/nNi=2)的化学腐蚀。
我们还采用前驱体浸渍焙烧法分别制备了ZnO和TiO2保护的0.5%-ZnO@NixP和0.5%-TiO2@NixP复合材料,以改善NixP(nP/nNi=2)在水环境中的稳定性。0.5% ZnO和0.5% TiO2包覆NixP(nP/nNi=2)样品的XRD如图9所示。复合材料的特征衍射峰均归属于六方晶相的Ni2P,表明少量金属氧化物在NixP(nP/nNi=2)表面的沉积并未改变内核NixP(nP/nNi=2)的晶体结构。复合样品的TEM测试表明,ZnO和TiO2保护层均匀地覆盖在NixP(nP/nNi=2)的表面(图10a和10e)。从HRTEM图可以看出,包覆ZnO和TiO2后,0.22 nm的晶格条纹间距源自NixP(nP/nNi=2)的(111)晶面,同时复合样品中均显示出清晰的ZnO-NixP和TiO2-NixP界面(图10b和 10f)。此外,HAADF-STEM 及EDX元素线扫图证明元素Zn、Ti、O、Ni、P均匀地分布在样品的表面(图10c、10d、10g、10h)。因此,上述表征结果证明通过简单的前驱体浸渍焙烧法成功制备了高质量的m-ZnO@NixP和n-TiO2@NixP复合材料。
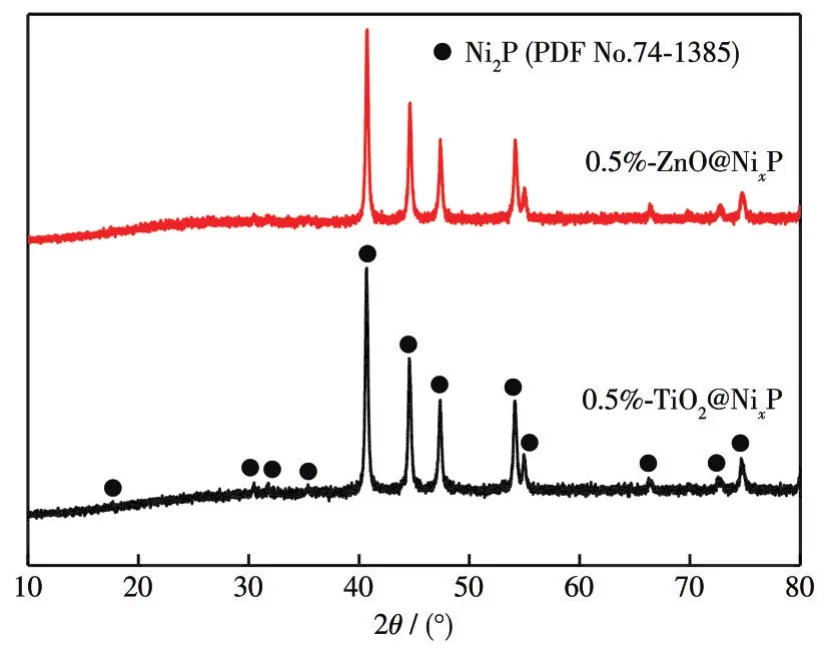
图9 0.5%-ZnO@NixP和0.5%-TiO2@NixP的XRD图Fig.9 XRD patterns of 0.5%-ZnO@NixP and 0.5%-TiO2@NixP
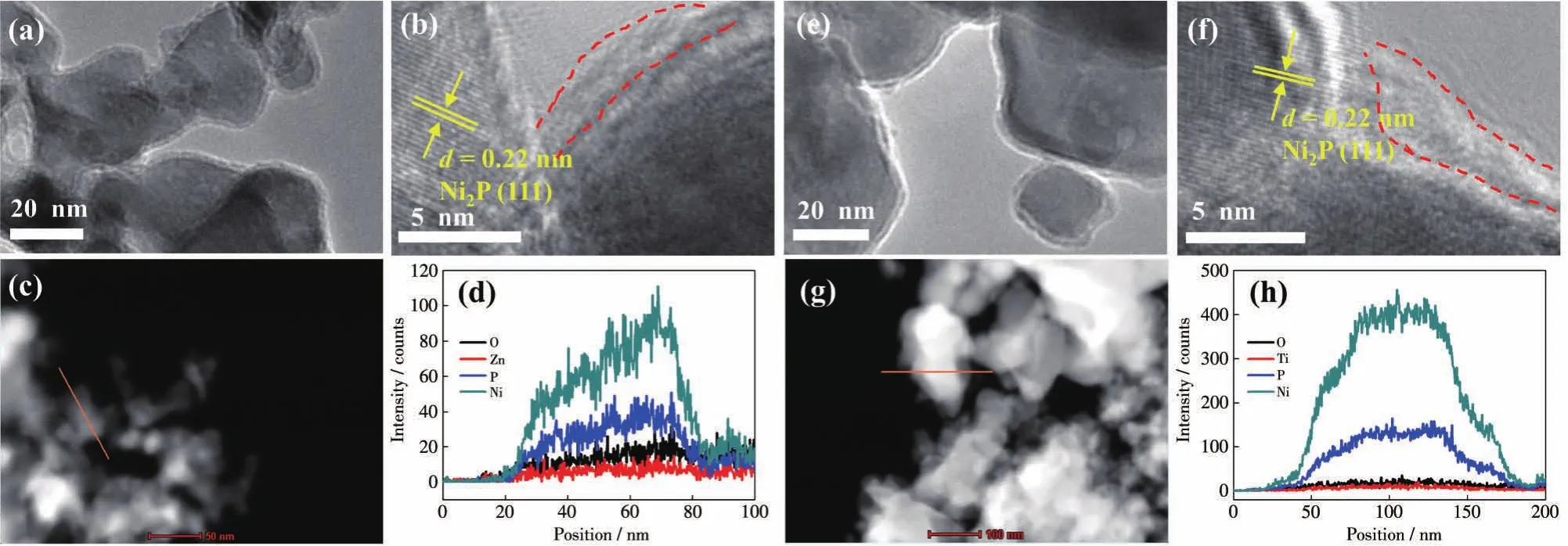
图10 0.5%-ZnO@NixP的(a)TEM图、(b)HRTEM图、(c)HAADF-STEM图和(d)元素线扫图;0.5%-TiO2@NixP的(e)TEM图、(f)HRTEM图、(g)HAADF-STEM图和(h)元素线扫图Fig.10 (a)TEM image,(b)HRTEM image,(c)HAADF-STEM image,and(d)element line scan curves of 0.5%-ZnO@NixP;(e)TEM image,(f)HRTEM image,(g)HAADF-STEM image,and(h)element line scan curves of 0.5%-TiO2@NixP
不同含量ZnO和TiO2包覆的m-ZnO@NixP和n-TiO2@NixP复合材料在水介质暗态条件下的析氢活性如图11a所示。显然NixP(nP/nNi=2)表面沉积的ZnO和TiO2保护层抑制了NixP(nP/nNi=2)在水中的腐蚀。对于m-ZnO@NixP体系而言,复合材料在水介质中的腐蚀析氢量随着ZnO覆盖比重的增加而降低;而在n-TiO2@NixP材料中,TiO2覆盖量为0.1%时即可显著抑制NixP(nP/nNi=2)的腐蚀,增加TiO2负载量对n-TiO2@NixP在水中析氢调控不明显。图11b是不同含量的ZnO和TiO2保护层构建的m-ZnO@NixP和n-TiO2@NixP复合材料在水中暗态反应3 h后Ni和P元素的浓度。相比于单一的NixP(nP/nNi=2)而言,复合材料反应溶液中的Ni2+和PO43-离子浓度大幅降低,这些结果证明惰性氧化物保护层能有效抑制NixP(nP/nNi=2)与水之间的化学腐蚀。
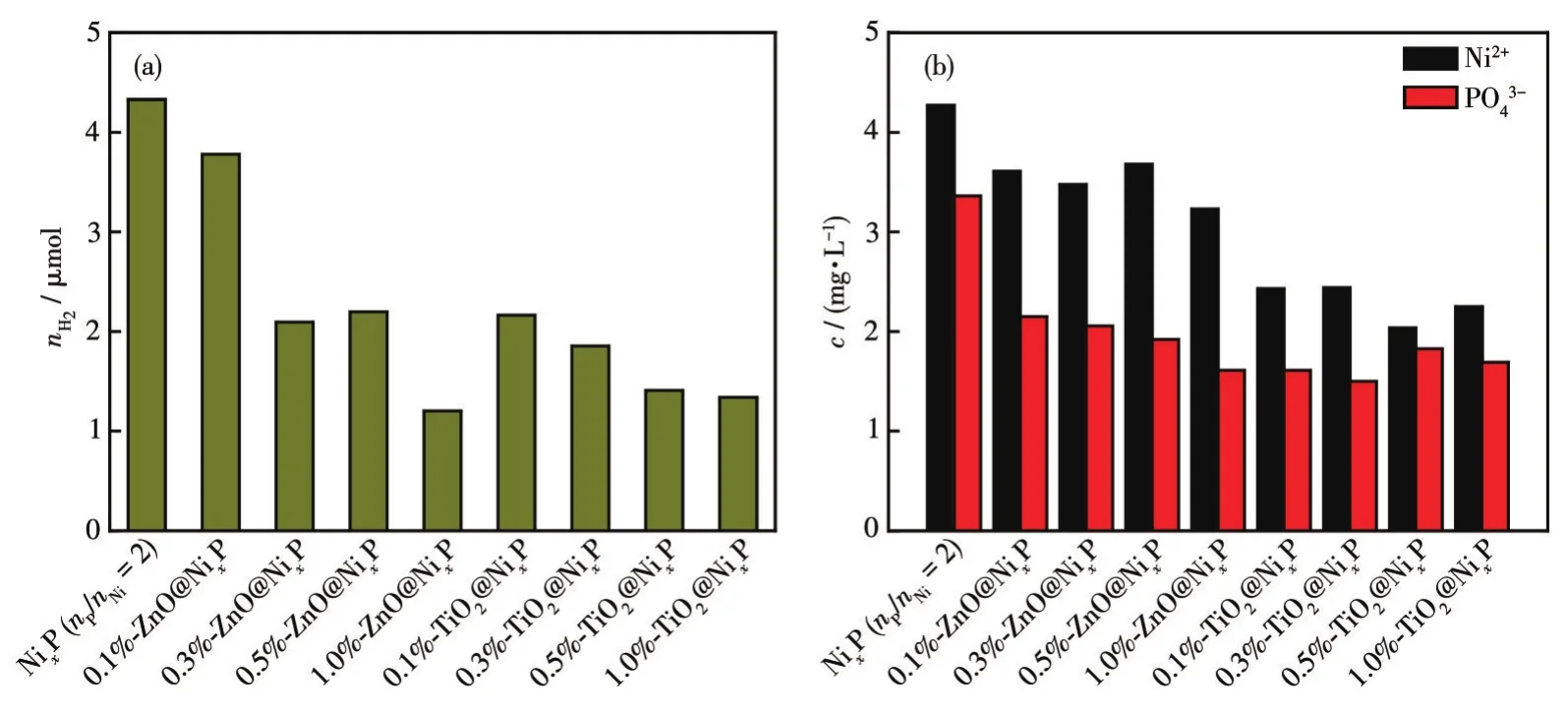
图11 m-ZnO@NixP和n-TiO2@NixP复合材料在水介质暗态条件下的(a)3 h反应后的析氢量和(b)3 h反应后溶液中的离子浓度Fig.11 (a)Hydrogen evolution activity and(b)ion concentration in the solution after 3 h reaction by m-ZnO@NixP and n-TiO2@NixP in the dark
2.4 NixP的腐蚀机制
基于上述的实验结果,NixP在水介质中的腐蚀过程包含P物种的氧化和水中质子的还原。如图12所示,当NixP置于水介质中时,水分子诱导Ni—P键断裂,其NixP表面的P物种失去电子被氧化为高价态的PO3-,同时析出的Ni2+与溶液中的OH-或PO3-44形成Ni(OH)2或Ni3(PO4)2,水中解离的H+得到电子被还原成H2[45]。与此同时,反应体系在光照条件下的析氢活性和PO43-浓度与暗态条件下相近,意味着NixP在水介质中的腐蚀与光照无关,其原因在于NixP的光激发载流子氧化还原电势较低[46],不足以驱动NixP和水的分解。
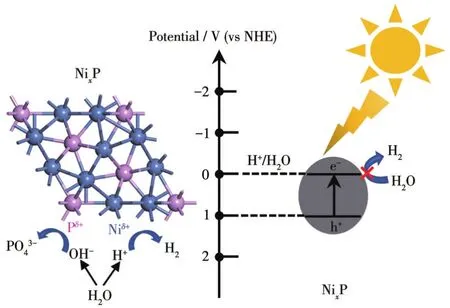
图12 NixP在水中的腐蚀机理Fig.12 Corrosion mechanism of NixP in water
3 结 论
NixP在中性水溶液中稳定性的研究结果表明,过渡金属磷化物NixP在暗态条件下即可与水反应生成H2,同时其表面的Ni和P因与水作用形成Ni2+和PO43-并溶于水中。不同nP/nNi时合成的NixP在水溶液中的腐蚀产氢与析出Ni2+、PO43-的行为不同。这是由于不同nP/nNi时合成的NixP化学组成、晶相、形貌和表面化学状态不同。光照并未明显加快NixP在水溶液中的腐蚀,表明NixP在水溶液中的腐蚀与光照无关。为了抑制NixP在水溶液中的腐蚀,可采用惰性氧化物包覆的方法,在NixP表面包覆惰性保护层NiO、ZnO和TiO2,经保护层覆盖后NixP在暗态条件下的产氢量和反应后溶液中Ni、P元素的浓度降低,表明其在水溶液中的腐蚀程度减小。
猜你喜欢
电镀与精饰(2022年10期)2022-10-14 08:37:12
社会科学战线(2022年3期)2022-06-15 02:43:58
金属加工(热加工)(2020年12期)2020-02-06 05:59:14
小学科学(学生版)(2018年1期)2018-01-31 01:51:37
电镀与环保(2017年6期)2018-01-30 08:33:37
电镀与环保(2017年5期)2017-12-19 12:06:05
电镀与环保(2017年3期)2017-06-23 08:24:51
中国煤炭(2016年9期)2016-06-15 20:29:53
采矿与岩层控制工程学报(2015年3期)2015-12-16 19:20:48
河南科技(2014年16期)2014-02-27 14:13:10

- 无机化学学报的其它文章
- La-Doped BaSnO3/Multi-walled Carbon Nanotube Modified Separator:Synthesis and Application in Lithium-Sulfur Battery
- Co(Ⅱ)/Ni(Ⅱ) Coordination Polymer of Isomeric Terphenyl-2,2″,4,4″-tetracarboxylic Acids with a Single Water Bridge:Syntheses,Structures,and Magnetic Properties
- Micromotors Based on Ni-Mn Binary Oxide and Its Application for Effective Dye Adsorption
- Direct Synthesis of Dimethyl Carbonate from CO2 and Methanol by Mg-Doped Ceria Monolithic Catalyst
- Hydrogen Storage Capabilities of the Low-Lying Ca2B4Clusters
- 盘状镝簇合物的合成及缓慢磁弛豫