
Hydrogen Storage Capabilities of the Low-Lying Ca2B4Clusters
2022-07-12 07:40TANGYuPengZHAOYanFeiYANGHaiYingLINan
无机化学学报 2022年7期
TANG Yu-PengZHAO Yan-FeiYANG Hai-YingLI Nan
(1Department of Applied chemistry,Yuncheng University,Yuncheng,Shanxi 044000,China)(2Science Experiment Center,Yuncheng University,Yuncheng,Shanxi 044000,China)(3State Key Laboratory of Explosion Science and Technology,School of Mechatronical Engineering,Beijing Institute of Technology,Beijing 100081,China)
Abstract:The structural feature and electronic property of Ca2B4,as well as its potential for hydrogen storage,have been studied using density functional theory.The first,second,and fourth low-lying isomers Ca2B401,Ca2B402,and Ca2B404 have high stabilities in thermodynamics and can adsorb 12,12,and 10 H2molecules with respective H2 gravimetric uptake capacity of 16.3%,16.3%,and 14.0%,which far exceeds the target(5.5%)proposed by the US department of energy(DOE).The average absorption energies per H2molecule are in the range of 0.58-4.21 eV for Ca2B401(H2)12,0.54-3.69 eV for Ca2B402(H2)12,and 0.10-0.12 eV for Ca2B404(H2)10.Born-Oppenheimer molecular dynamic(BOMD)simulations indicate Ca2B401 and Ca2B402 are promising candidates for adsorbing hydrogen,but Ca2B404 is not.The results of hydrogen adsorption energies with Gibbs free energy correction indicate that 12 H2 molecules on Ca2B401 and Ca2B402 are energetically favorable with a wide range of temperatures at 101 325 Pa.
Keywords:hydrogen storage;density functional theory;absorption;molecular dynamic;Gibbs free energy
Hydrogen is considered to be a sustainable and eco-friendly energy carrier because of its abundance,easy synthesis,and high heat thermal capacity on the earth[1-2].However,the wide-scale use of hydrogen fuel hinges on our ability to find safe and cost-effective hydrogen storage materials.The ideal hydrogen storage materials should meet the stringent requirements:high gravimetric and volumetric density,fast kinetics,and thermodynamics that allow reversible hydrogen adsorption and desorption in H2molecular form to take place under ambient conditions[3-6].According to the guidelines set by the US Department of Energy(DOE),a minimum requirement for a system to be a potential hydrogen storage candidate is that it should possess a minimum H2gravimetric uptake capacity of 5.5% and delivery under 1 200 kPa pressure in the operating ambient temperature range of 233 to 333 K[7].
Traditionally,the storage materialsbind the hydrogen atoms primarily through three different processes[1,6].In chemisorption,the H2molecules dissociate into individual atoms,migrate into the storage material,and are strongly bonded with the binding energy in the range of 2-4 eV like chemical hydrides[8-9],in which the strong interaction makes it difficult to release H2during application.On the other hand,like the pure carbon-based nanostructures,the H2is bonded weakly via physisorption and remains in its molecular form with the binding energy in the range of few meV[10].However,the major drawbacks in physisorption are that the adsorption must be carried out at a very low temperature and high gas pressure.Recently,more attempts have been made to design and develop new hydrogen storage materials based on the third form of adsorption,which is intermediate between physisorption and chemisorption with the binding energy of 0.1-0.8 eV and considered to be essential for the faster adsorption and desorption kinetics for vehicular application.It includes metal-decorated nanomaterials[11-18],transition metal-acetylene/ethylene[19-28],and transition metal clusters[29-33].For example,Sun et al.[14]predicted the hydrogen storage capacities of the Li12C60cluster in which each Li atom could adsorb a maximum of 5 H2molecules leading to a gravimetric density(w/w)of 13%.Durgun et al.[23]theoretically indicated Ti2-C2H4could adsorb a maximum of 10 H2molecules with the average binding energy of 0.45 eV.Du et al.[29]recently predicted that the carbon motif CTi72+could bind 20 H2molecules at most,which resulted in a gravimetric density of 19%.
Compared with carbon-based materials,metaldecorate boron clusters have also been considered a promising candidate for hydrogen storage[34-42].For example,B6Li8was predicted to be an excellent hydrogen storage media with gravimetric density likely reaching up to a theoretical limit of 24%[35].Du et al.[39]have investigated the hydrogen storage capacity of the Saturn-like charge-transfer complex Li4B40,in which each Li atom could bind 6 H2molecules at most resulting in the gravimetric density of 10.4%.Just like the alkali metal decorated materials,boron clusters doped by transition metals have become a research hotspot[43-47].Very recently,the highly stable Sc2B42+cluster was investigated as a promising candidate for hydrogen storage material,which corresponded to a hydrogen uptake of 17.49% and average binding energy of 0.42 eV[46].As we know,the transition metal Sc is expensive and charge neutrality should be a consideration in the engineering of practical materials for hydrogen storage.On the other hand,we have called attention to the isoelectronic relationship of a Ca atom to a Sc+ion.Moreover,calcium has been suggested to functionalize the nanomaterials as hydrogen storage materials because of its relatively small cohesive energy and moderate interaction with H2molecules[15-16,40-41,48].For example,the inverse sandwich Ca2B8was found to be a promising hydrogen storage material that showed moderate adsorption energy and high gravimetric density(10.6%)for H2[48].
Therefore,in the current work,we choose Ca2B4as the theoretical research model to investigate the corresponding geometrical configuration and electronic structures,and further probe into the hydrogen storage abilities of the low-lying isomers.
1 Computational methods
The 1 000 initial structures of Ca2B4were generated by a stochastic search method embedded in the Molclus program[49],and the resulting structures were optimized in the singlet state and triplet state at the PBE0/6-311+G(d)level[50-53],respectively.The PBE0 functional is an effective tool in studies of the metaldoped boron clusters[54-55].The vibrational frequencies of all the local minima were confirmed at the same level to guarantee that the structures optimized are true minima on the potential energy surface.To obtain the reasonable adsorption energy of H2molecules on Ca2B4clusters,the molecular structures of the isolated and H2-adsorbed Ca2B4were further fully optimized without any symmetry constraints using the ωB97XD functional[56]in conjunctions with 6-311+G(d,p)basis set.The ωB97XD functional with the long-range interactions has been proven to be an authentic method for predicting non-covalent interactions[29,39,42-43].The basis set superposition errors(BSSE)[57]were corrected using the full counterpoise method for all the H2-adsorbed Ca2B4structures.To evaluate the reversibility of storage of H2molecules,the successive adsorption energy(ΔEs)and the average absorption energy per H2molecule(ΔEa)were calculated at ωB97XD/6-311+G(d,p)level according to the following formulas:

WhereEXstands for the total energy of X(X=Ca2B4,H2,Ca2B4(H2)n-1,Ca2B4(H2)n).Notably,the spontaneous adsorption of H2can occur if the ΔEsis positive,and the negative ΔEsmeans the successive adsorption is difficult.
The H2gravimetric density of Ca2B4was calculated using the following equation:
Gravimetric density=MH2/(MH2+MCa2B4)×100% (3)WhereMH2represents the mass of the total number of H2molecules adsorbed andMCa2B4represents the mass of the host Ca2B4.
Besides,Born-Oppenheimer molecular dynamics(BOMD)simulations at the temperatures of 77 and 300 K were performed for the relaxed structures of selected species Ca2B401(H2)12,Ca2B402(H2)12,and Ca2B404(H2)10at the ωB97XD/6-31+G(d,p)level.
All the geometry optimization and property calculation were performed using the Gaussian 09 package.
2 Result and discussion
2.1 Geometrical and electronic structure of Ca2B4
2.1.1 Geometrical structure of Ca2B4
A total of 16 low-lying isomers for Ca2B4were identified via extensive structural searches.To ensure the energetics,all optimized isomers were benchmarked using single-point CCSD(T)[58]calculations.Fig.1 represents the structures and relative energies of each isomer at the CCSD(T)/6-311+G(d)//PBE0/6-311+G(d)level.Ca2B401 is the most stable structure,which is only 0.035,0.078,and 0.089 eV less in energy than the top three competitors(Ca2B402,Ca2B403,and Ca2B404),respectively.On the other hand,although Ca2B402 and Ca2B403 have the same geometries,Ca2B402 with a singlet state is more stable than the triplet Ca2B403.Therefore,the geometrical configurations,electronic properties,and the hydrogen storage abilities for the first,second,and fourth low-lying isomers Ca2B401,Ca2B402,and Ca2B404 were researched.The calculated key bond lengths of the bare and H2adsorbed compounds Ca2B401,Ca2B402,and Ca2B404 at ωB97XD/6-311+G(d,p)level are listed in Table 1.Notably,Ca2B401(H2)12and Ca2B402(H2)12have the same geometries.Comparing the isolated Ca2B4isomers,the corresponding B—B and B—Ca bonds in Ca2B402(H2)12and Ca2B404(H2)10are not considerable change,which indicates the structures of Ca2B4are not distorted with adsorption of a maximum of H2molecules.Moreover,the smallest vibrational frequency(Table 1)of the bare isomers is predicted to be 98,115,and 130 cm-1,which are sufficiently large to meet a stability criterion suggested by Hoffmann et al.[59].In addition,the energy gap(ΔEHL)between the highest occupied molecular orbital(HOMO)and lowest unoccupied molecular orbital(LUMO)was also calculated to analyze the stabilities of Ca2B401,Ca2B402,and Ca2B404 due to a large ΔEHLcan reflect the high stabilities of compounds.The ΔEHLof Ca2B401,Ca2B402,and Ca2B404 are 4.02,4.16,and 4.88 eV,respectively,indicating the three clusters have high stabilities.To further study the thermodynamic stabilities,BOMD simulations were carried out for 5 ps at 300 K.As shown in Fig.2,the relative potential energies for Ca2B401,Ca2B402,and Ca2B404 in the simulated time show slight oscillations,suggesting their high stabilities at room temperature.To gain clear geometries,the extracted snapshots of Ca2B401,Ca2B402,and Ca2B404 at different simulation times(50,2 500,and 5 000 fs)are also depicted in Fig.2.

Fig.1 Optimized low-lying isomeric structures of Ca2B4at PBE0/6-311+G(d)level

Fig.2 Variations of potential energy vs simulation time at 300 K for Ca2B401,Ca2B402,and Ca2B404
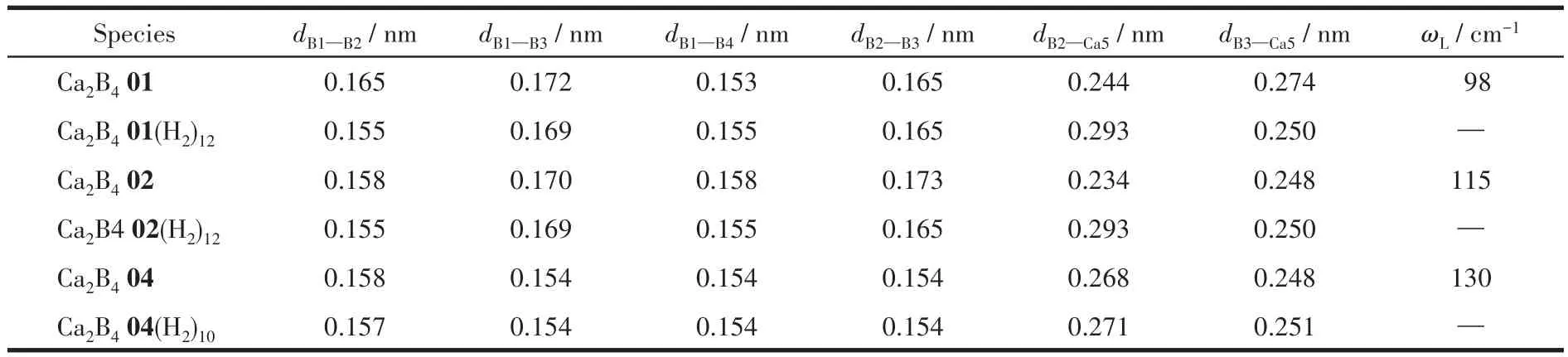
Table 1 B—B/Ca bond distances(nm)and the lowest vibrational frequency ωL(cm-1)of the isolated and multiple H2adsorbed compounds at ωB97XD/6-311+G(d,p)level
2.1.2 Electronic structure of Ca2B4
To analyze the electronic structure and the effect of H2molecules adsorbed,the Mulliken charge for Ca2B4isomer has been calculated at ωB97XD/6-311G(d,p)level.As shown in Fig.3,in Ca2B401,the boron and calcium atoms carry about-0.183e,-0.651e,-0.181e,-0.092e,0.554e,and 0.555e,respectively.For Ca2B402,the four boron atoms(B1-B4)have-0.283e,-0.314e,-0.586e,and-0.314e,respectively.Two calcium atoms(Ca5-Ca6)have 0.749e and 0.749e,respectively.In Ca2B404,the boron and calcium atoms carry about-0.234e,-0.234e,-0.646e,-0.646e,0.880e,and 0.880e,respectively.The strong charges transfer from calcium atoms to boron atoms when these compounds are formed.Therefore,partially charged Ca ion and B4 may produce a local electrostatic field that can polarize H2molecules and then bind them via the polarization mechanism.To further illustrate the above concept,the contour plots of the molecular electrostatic potential(ESP)of Ca2B401,Ca2B402,and Ca2B404 isomers were also obtained by Multiwfn[60].As illustrated in Fig.4,the calcium and boron atoms have a positive and negative potential,respectively.The map ofESPdiffusion accords with the Mulliken charge analysis,indicating the H2molecules should be preferentially ad-sorbed on calcium atoms.

Fig.3 Mulliken charge of Ca2B401,Ca2B402,and Ca2B404 at the ωB97XD/6-311G(d,p)level
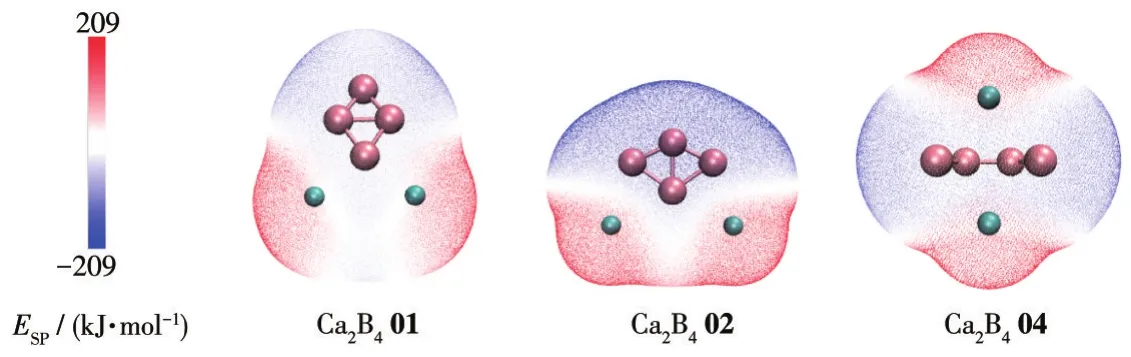
Fig.4 ESPmaps of Ca2B401,Ca2B402,and Ca2B404 at the ωB97XD/6-311G(d,p)level
2.2 H2adsorption behavior of Ca2B4
2.2.1 H2adsorption behavior of Ca2B401
We next studied the sequential hydrogenation of Ca2B401.Based on the above analysis,the Ca atom is the most active atom in all sites during the process of H2adsorption.Considering the symmetry of the isomers Ca2B401,a number of H2molecules were successively placed around every Ca atom,and the structures were optimized without any symmetry constraints at the ωB97XD level of theory,respectively.The optimized structures of the isomer Ca2B401 with adsorbed multiple H2molecules at the ωB97XD level of theory are depicted in Fig.S1 (Supporting information).The selected relaxed configurations Ca2B401(H2)12is depicted in Fig.5.
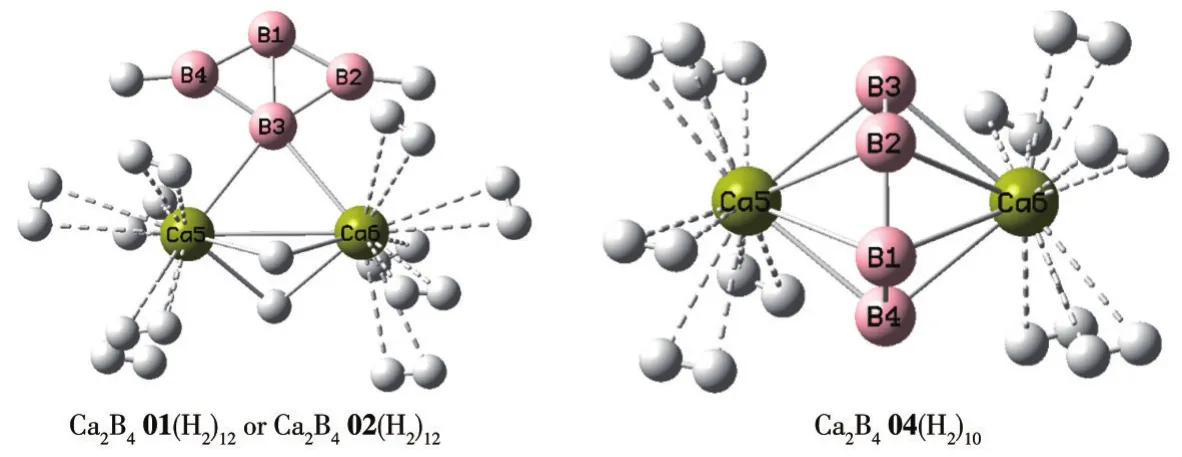
Fig.5 Optimized geometries of Ca2B401(H2)12,Ca2B402(H2)12,and Ca2B404(H2)10at the ωB97XD/6-311+G(d,p)level
Ca2B401 can at most adsorb 12 H2molecules and the gravimetric density of stored hydrogen is 16.3%,which is about 3.3 times larger than the criteria of 5.5% proposed by DOE[7].As listed in Table 2,the bond lengths of the adsorbed molecular form H2in Ca2B401(H2)12are 0.075 nm,which is elongated compared to the bond length(0.074 nm)of isolated H2at the same calculated level.Notably,the binding of the first molecule to Ca2B401 isomer has five different characteristics(Fig.S1).Among them,the H2molecule interacts dissociatively with two B atoms and the resulting borohydride structure is the ground-state(1a),which is 0.35,0.37,0.46,and 4.21 eV lower in energy than 1b,1c,1d,and 1e,respectively.It is for this reason that we have concentrated an adding successive H2molecules to the ground-state structure.Next,the adsorption of the second H2molecule to Ca2B401(H2)1(1a)has two different characteristics.Here,hydrogen binds molecularly to one of the Ca atoms and the resulting isomer(Fig.S1,2b)is 0.72 eV higher in energy than the ground-state structure(2a)in which the second H2molecule dissociates with two H atoms bridging between two Ca atoms.As listed in Table 3,the successive binding energies of the ground states of Ca2B401(H2)1and Ca2B401(H2)2are 4.21 and 1.67 eV respectively,indicating the binding of the first two molecules belongs to the chemisorption process.It is only when the third H2molecule is added to 2a that the binding becomes molecular,with successive energy of 0.11 eV.The H2molecules from the fourth to the twelfth also bind to the Ca atoms in nearly molecular form.The binding energies of each successive H2molecule are in the range of 0.11-0.12 eV as one proceeds from Ca2B401(H2)3to Ca2B401(H2)12.To confirm the adsorption of H2molecules to Ca2B401 is reversible or not,the ΔEaof Ca2B401(H2)n(n=1-12)are also illustrated in Table 3.Ca2B401 adsorbs 1-12 H2molecules with the ΔEaof 4.21 to 0.58 eV.It can be found that some ΔEavalues are too large and exceed the energy criteria(0.1-0.8 eV)of the reversible hydrogen storage.Because the strong chemical bonds between the dissociation of the first two H2molecules and B/Ca atoms improve the ΔEaof compounds.However,more remarkably,the average adsorptions energy of Ca2B401(H2)12is 0.58 eV,which is ideal for reversible hydrogen storage at near ambient conditions.

Table 2 Ca—H and corresponding H—H distances(nm)of Ca2B401(H2)12/Ca2B402(H2)12and Ca2B404(H2)10at ωB97XD/6-311+G(d,p)level

Table 3 Calculated ΔEsand ΔEawith BSSE correction and without zero-point energy correction at ωB97XD/6-311+G(d,p)level
2.2.2 H2adsorption behavior of Ca2B402
The optimized structures of H2-adsorbed Ca2B402 are depicted in Fig.S2.Notably,the binding of the first molecule to Ca2B401 isomer has three different characteristics(Fig.S2)and the ground-state structure of Ca2B402(H2)1(1a′)is exactly the same as Ca2B401(H2)1(1a).As illustrated in Table 3,the successive binding energies of the ground state Ca2B401(H2)1(1a′)is 3.69 eV,indicating the binding of the first molecule belongs to the chemisorption process.It is when the second H2molecule is added to 1a′that the H2adsorbed structures Ca2B402 are exactly the same as the corresponding H2adsorbed structures Ca2B401.The ΔEaof Ca2B402(H2)n(n=1-12)are also listed in Table 3.Ca2B402 adsorbs 1-12 H2molecules with the ΔEaof 3.69 to 0.54 eV.The ΔEaof Ca2B402(H2)12is 0.54 eV,which is ideal for reversible hydrogen storage at near ambient conditions.
2.2.3 H2adsorption behavior of Ca2B404
The optimized structures of H2-adsorbed Ca2B404 are depicted in Fig.S2.Ca2B404 can successively adsorb 10 H2molecules in total,from one to five H2molecules on each Ca atom.The selected relaxed configuration Ca2B404(H2)10is depicted in Fig.5.The Ca—H distances and the corresponding H—H bond lengths are listed in Table 2.The Ca—H distances are in a range of 0.235-0.267 nm,and the corresponding H—H bond lengths are elongated to 0.076-0.077 nm.As shown in Table 3,the ΔEsvalues are in a range of 0.10-0.14 eV for H2-adsorbed Ca2B404.The positive energy values of ΔEsindicate that 10 H2molecules can be effectively adsorbed on Ca2B404.Besides,the ΔEaof Ca2B404(H2)n(n=1-10)are in a range of 0.10 to 0.12 eV which meets the criteria(0.1-0.8 eV)of reversible hydrogen storage.For Ca2B404(H2)10,the gravimetric density of stored hydrogen is 14.0%.The result surpasses the target for hydrogen uptake capacity specified by DOE.
2.3 Reversibility of H2molecules on Ca2B401,Ca2B402,and Ca2B404
To test the hydrogen release for Ca2B401(H2)12,Ca2B402(H2)12,and Ca2B404(H2)10at ambient conditions,we performed the BOMD simulations at the ωB97XD/6-31+G(d,p)level.The BOMD simulations were carried out with a time of scale of 800 fs with a trajectory step size of 0.5 fs at the temperatures of 77 and 298 K.Fig.6 shows the potential energies as functions of time,and the extracted snapshots at different simulation times(50,100,200,300,400,and 500 fs)are depicted in Fig.S3-S6.For Ca2B401(H2)12/Ca2B402(H2)12,10 H2molecules far from Ca sites have begun to run away from the host Ca2B401 cluster within 100 fs,and the H2molecules desorb faster at the higher temperatures.On the other hand,at the end of simulations of 77 and 298 K,only the first two H2molecules are still adsorbed in atom form,whereas the other ten H2in the molecular form completely escape from Ca2B401,corresponding to a release ratio of 83.3%,which is excellent agreement with the discussed adsorption mechanism above.Interestingly,Ca2B401(H2)2structure shows a high dynamic stability at 77 and 298 K,being in line with the values of ΔEaand ΔEs.Moreover,it can be found from the snapshots of Fig.S3 and S4 that the host clusters Ca2B401 and Ca2B402 are not significantly deformed during the dynamic simulation processes.Therefore,we can conclude that the clusters Ca2B401 and Ca2B402 are appropriate candidates for reversible hydrogen storage.For Ca2B404(H2)10,at the processes of simulations of 77 and 298 K,although most of the H2adsorbed can also completely escape from Ca2B404,the host cluster Ca2B404 is significantly deformed.Thus,Ca2B404 is not an appropriate candidate for reversible hydrogen storage.

Fig.6 Potential energy trajectories of(a)Ca2B401(H2)12/Ca2B402(H2)12and(b)Ca2B404(H2)10complexes at the temperatures of 77 and 298 K
2.4 Gibbs free energy corrected adsorption energies(ΔEG)
To confirm the adsorptions of Ca2B401(H2)12and Ca2B402(H2)12are favorable or not at different temperatures,the ΔEGwas calculated at different temperatures and 101 325 Pa.The formula isare the calculated Gibbs free energies of the bare cluster Ca2B4,H2molecule,and Ca2B4(H2)12,respectively.The positive ΔEGvalue reflects that the adsorption of multiple H2molecules on Ca2B4is energetically favorable at the corresponding condition.As shown in Fig.7,the ΔEGof Ca2B401(H2)12and Ca2B402(H2)12are still positive at 400 K at 101 325 Pa.It indicates that both Ca2B401(H2)12and Ca2B402(H2)12have fairly wide temperature ranges on which we can tune the thermodynamically favorable hydrogen adsorption just near room temperature at 101 325 Pa.

Fig.7 Temperature dependence of ΔEGvalues for Ca2B401(H2)12and Ca2B402(H2)12at ωB97XD/6-311+G(d,p)level
3 Conclusions
In this work,the structures,stabilities,and hydrogen storage behavior of Ca2B4have been researched using density functional theory.According to the calculations,the first,second,and fourth low-lying isomers Ca2B401,Ca2B402,and Ca2B404 have high stabilities in thermodynamics at 300 K.For Ca2B401 and Ca2B402,the resulting H2adsorbed structures are the same and up to 12 H2molecules can be bound.Ca2B404 can adsorb 10 H2molecules at most.The systems can have a maximum gravimetric density of 16.3% and 14.0% for Ca2B401(H2)12/Ca2B402(H2)12and Ca2B404(H2)12,respectively,which satisfy the target specified by US DOE.The ΔEaof 0.58-4.21 eV for Ca2B401(H2)12,0.54-3.69 eV for Ca2B402(H2)12,and 0.10-0.12 eV for Ca2B404(H2)10are in the range of the physisorption and chemisorption energy.The results of BOMD simulations indicate Ca2B401 and Ca2B402 can be promising candidates for adsorbing dihydrogen,but Ca2B404 is not.Moreover,the temperature-dependent Gibbs free energy corrected adsorption energies indicate Ca2B401 and Ca2B402 are suitable for storage H2with a wide range of temperatures at 101 325 Pa.
Supporting information is available at http://www.wjhxxb.cn
