
Structural requirements and interaction mechanisms of ACE inhibitory peptides: molecular simulation and thermodynamics studies on LAPYK and its modified peptides
2022-07-11 05:49BiyingZhngJingboLiuHediWenFengJingErleiWngTingZhng
食品科学与人类健康(英文) 2022年6期
Biying Zhng, Jingbo Liu, Hedi Wen, Feng Jing, Erlei Wng, Ting Zhng,*
a Jilin Provincial Key Laboratory of Nutrition and Functional Food, College of Food Science and Engineering, Jilin University, Changchun 130062, China
b College of Food Science and Engineering, Jilin Agricultural University, Changchun 130118, China
ABSTRACT
The understanding of the structural requirements and the intermolecular-interaction mechanism are important for discovering potent angiotensin-converting enzyme (ACE) inhibitory peptides. In this study, we modified an egg-white derived peptide, LAPYK, using the amino acids with different properties to produce the LAPYK-modified peptides. The ACE inhibitory activities of the modified peptides were determined to explore the structural requirements of ACE inhibitory peptides (ACEIPs). Molecular simulation and isothermal titration calorimetry analysis were used to investigate interactions between the peptides and ACE.We found that hydrophobicity and the amino acids with ring structures were beneficial for the ACE inhibitory activities of the peptides. The results of the molecular mechanics poisson boltzmann surface area (MMPBSA)binding free energy calculations indicated that the polar solvation free energy (ΔGpolar) of the charged peptides (LAPYK, LAPYE) were unfavorable for binding to ACE. On the other hand, the results of isothermal titration calorimetry analyses suggested that the enthalpy-driven ACE-peptide interactions were more favorable than the entropy-driven ACE-peptide interaction counterparts.
Keywords:
ACE inhibitory peptides
Molecular docking
Molecular dynamics simulation
Isothermal titration calorimetry
1. Introduction
The angiotensin I-converting enzyme (ACE) is a zinc metallopeptidase that plays a vital role in the renin-angiotensin-aldosterone system (RAS) [1]. ACE converts Angiotensin I to Angiotensin II and increases blood pressure [2]. Thus, inhibiting the activity of ACE is an effective way to lower blood pressure. Commercially antihypertensive drugs, such as Captopril [3] and Lisinopril [4],are the inhibitors for ACE activity. However, these synthetic drugs have some undesirable side effects [5]. Food-derived peptides are expected to be a safer alternative due to their lower side effects [6]. In recent years, researchers have identified the ACE inhibitory peptides (ACEIPs) derived from food such as egg [7], casein [8], and meat [9].
The ACE inhibitory activities are related to the structural conformations of peptides [10]. To explore the structural-activity relationships of ACEIPs, researchers have established QSAR models. Wu et al. [11] established the models of dipeptides and tripeptides using 3 Z-scores of 20 coded amino acids. Guan et al. [12]established the QSAR model of ACEIPs using the amino acid descriptor SVHEHS. According to these models, the relationships between the peptide structural and ACE inhibitory activity were concluded. For example, Shu et al. [13] reported that hydrophobic and steric properties are important for the ACE inhibitory activities of peptides. Liu et al. [14] suggested the importance of aromatic amino acids for the ACE inhibitory activities of peptides. The reports on the existing QSAR models suggest that the modifications of previously reported ACEIPs enhance the current understandings of the structural requirements of ACEIPs for improving the ACE inhibitory activities of the peptides.
The analyses of protein-ligand interactions complement the structural requirements analyses to elucidate the inhibitory mechanism of ACEIPs. Molecular docking is a common technique to predict the binding sites and the molecular interactions of ACEIPs [15]. Yu et al. [16]found that the ACEIP TNGIIR interacts with the S1 S2, and S1’pockets of ACE. Fan et al. [17] reported that the peptides that exhibit high ACE inhibitory activities form a large number of hydrogen bonds with ACE. Molecular dynamics (MD) simulation is an alternative to molecular docking; it is an essential tool to analyze protein-ligand interactions, to characterize the protein-ligand complex dynamic behaviors, and to calculate the free energies of binding by the molecular mechanics poisson boltzmann surface area (MMPBSA)method [18]. Recently, MD simulation has been incorporated in the discovery of the drugs for SARS-CoV-2 [19]. Thermodynamics parameters are the other invaluable indicators for the analyses of protein-ligand interactions, and isothermal titration calorimetry (ITC)is an effective method to obtain thermodynamics information about the ACE-peptide binding [20]. However, the applications of MD simulation and ITC in studies involving ACEIPs are limited.
The understanding of the structural requirements and the interaction mechanism are important to improve the efficacies of ACEIPs. In this study, we used the peptide LAPYK as a template and modified LAPYK by replacing the C-terminal lysine (K) residue with different amino acids. The ACE inhibitory activities of the modified peptides were determined to understand the structural requirements of ACE inhibitory peptides. For further, the molecular simulation and ITC analyses were used to understand the intermolecular interactions between ACE and the peptides.
2. Materials and methods
2.1 Materials
Abz-Gly-Phe(NO2)-Pro (Abz) was purchased from Bachem Feinchemikalien (Bubendorf, Switzerland). ACE (9015-82-1,≥ 2.0 units/mg) was purchased from Sigma-Aldrich Company (St.Louis, MO, USA). The peptides (≥ 98%) were synthesized using the Fmoc solid phase method by Sangon Biotech Co., Ltd. (Shanghai,China). Tris (hydroxymethyl) methyl aminomethane, NaCl, and NaOH were analytically pure.
2.2 The properties of the peptides
The hydrophobic properties of amino acids were normalized by following the procedures described by Monera et al. [21]. The charges of the amino acids were deduced from the amino acid reference table provided by Merk (https://www.sigmaaldrich.com/china-mainland/lifescience/metabolomics/learning-center/amino-acid-reference-chart.html). The hydrophobic properties and the charges of peptides were obtained from the Property Calculation of Antimicrobial Peptide Database (https://dbaasp.org/property-calculation).
2.3 The determinations of ACE inhibitory activities
The ACE inhibitory activities of the peptides were determined using SHIMADZU 2030C 3D series Rapid Resolution LC System(Shimadzu, Kyoto, Japan) according to the method reported by Sentandreu and Toldrá [22] with some modifications. Briefly, the substrate Abz (450 μmol/L) was dissolved in Tris buffer (pH 8.3)and added with 1.125 NaCl. Peptide solution (50 μL) was added to 200 μL of the substrate solution and the mixture was pre-incubated at 37 °C. 50 μL of ACE solution (5 mU/mL) was added to the pre-incubated solution to begin the reaction. The generated fluorescence was measured at 0 and 30 min. The excitation and emission wavelengths were 355 and 420 nm, respectively.
2.4 Molecular docking analyses
The molecular dockings between the peptides and ACE were analyzed by Autodock 4.2 [23] according to our previous study [24].The structure of ACE (PDB ID: 1O86) was obtained from the RCSB Protein Data Bank (http://www.rcsb.org). The size of the grid box was 100 Å × 100 Å × 100 Å and the grid spacing was 0.375 Å. The coordinates of zinc ion (x: 43.821, y: 38.240, z: 46.712) were set to the center of the grid box. A total of 200 runs were performed to discover the proper docking poses between the peptides and ACE.
2.5 MD simulation
The MD simulations of ACE-peptides complexes were performed using Gromacs [25]. The box was cubic-shaped and the minimum distance of any protein atom to the closest box wall was 1 nm. The system energy was minimized by the steepest descent method until the energy was less than 1 000 kJ/(mol·nm). Then, the system was pre-balanced under the NVT ensemble and the NPT ensemble.Finally, the simulations were performed for 200 ns with a time step of 2 fs. During the simulation, the H-bonds were constrained using the LINCS algorithm [26]. The electrostatic interactions among non-ligand atoms were evaluated by the PME (Particle Mesh Ewald)method [27]. The temperature coupling was implemented by the Nose-Hoover algorithm [28]. The pressure coupling was implemented by the Parrinello-Rahman algorithm [29].
2.6 The calculations of the binding free energy
The MMPBSA calculations of the binding free energy were performed using gmx_mmpbsa script (https://github.com/Jerkwin/gmxtool/blob/master/gmx_mmpbsa/gmx_mmpbsa.bsh) based on the following equation:
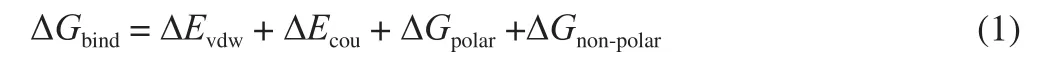
where the ΔEvdwand ΔEcourepresented the electrostatic energy and the van der Waals energy in a vacuum. ΔGpolarand ΔGnon-polarrepresented the polar and non-polar solvation energy.
2.7 Isothermal titration calorimetry (ITC) analyses
The ITC experiments were performed by an iTC200 microcalorimeter (GE, Fairfield, CT, USA). ACE solution (119 nmol/L,190 μL) dissolved in Tris buffer (pH 8.3) was added into the sample cell. The reference cell was filled with Tris buffer (pH 8.3). The peptides solution (0.1 mmol/L, 40 μL) was added into the syringe. All of the experiments were performed at 25 °C. After the baseline had been stable, the peptides were titrated via 18 times of 2 μL individual injections with a 180 s intervals. The stirring speed was 1 000 r/min.
2.8 Statistical analysis
The activity tests were performed as three independent measurements and the data were expressed as mean ± standard deviations (SD). The independent sample t-test was selected to analyze the significant differences in the ACE inhibitory activities among the peptides.
3. Results and discussion
3.1 The properties of peptides
Charge, hydrophobicity, and steric of the peptides were reported to be relative to their ACE inhibitory activities [30,31]. Besides, the potent ACE inhibitory activities of the peptides IPP and VPP suggest that proline might be suitable for the ACEIPs. To explore the effect of the C-terminal amino acids on the ACE inhibitory activities of peptides, we substituted the C-terminal lysine of the peptide LAPYK with phenylalanine (hydrophobicity), proline, tryptophan (steric), and glutamic acid (charge) and obtained the peptides LAPYF, LAPYP,LAPYW, and LAPYE. Subsequently, we exchanged the N/C terminal amino acids of the peptides and obtainedfive new peptides (KAPYL,FAPYL, PAPYL, WAPYL, and EAPYL). The properties of the peptides and the N/C-terminal amino acids are shown in Fig. 1.

Fig. 1 The hydrophobicity scales of the modified peptides and their N/C-terminal amino acids.
The charges of the peptides have been reported to in fluence the ACE inhibitory activities of the peptides [30]. The peptide LAPYK had a positive charge due to the positively charged lysine (K) side chain. To investigate the effects of the charges of the C-terminal amino acids, we substituted the lysine with the negatively charged glutamic acid and the amino acids with zero overall charges.Therefore, we investigated the ACE inhibitory activities of the 2 peptides that have the positive charge, which were LAPYK and KAPYL, the 2 peptides that have the negative charge, which were LAPYE and EAPYL, and 6 peptides that have zero charge.
Hydrophobicity is necessary for the activities of ACEIPs [31].The hydrophobic amino acids at both the N-terminus [32] and C-terminus [33] are beneficial for the ACE inhibitory activities of peptides. As shown in Fig. 1, the order of the amino acids on the basis of hydrophobicity is phenylalanine (F) > tryptophan (W) =leucine (L) > lysine (K) > glutamic acid (E) > proline (P). On the other hand, the order of the peptides on the basis of hydrophobicity is LAPYF (FAPYL) > LAPYP (PAPYL) > LAPYW (WAPYL) >LAPYE (EAPYL) > LAPYK (KAPYL). It’s interesting to note that the hydrophobicity order of the peptides did not correspond to that of the amino acids, although only terminal amino acid was replaced.
3.2 The ACE inhibitory activities of modified peptides
The ACE inhibitory activities of modified peptides are presented in Fig. 2. The results showed that the ACE inhibitory activity of the peptide LAPYE (IC50> 1 000 μmol/L) was lower than others. The same results were obtained after the exchange of N/C-terminal amino acids. These findings indicated that the peptides with zero charge exhibited higher ACE inhibitory activities than did the peptides with a negative charge. Ourfindings contradicted thefindings reported by Aluko [34]. The authors suggested that negatively charged peptides may interact with the zinc ion of ACE and reduce the catalytic activity of ACE. The plausible explanation for the difference in ourfindings and the findings by Aluko [34] might be that the LAPYE was not able to enter the center of the hydrophobic cavity of ACE at which the zinc ion was stabilized; thus, the peptide was not able to interact with the zinc ion.
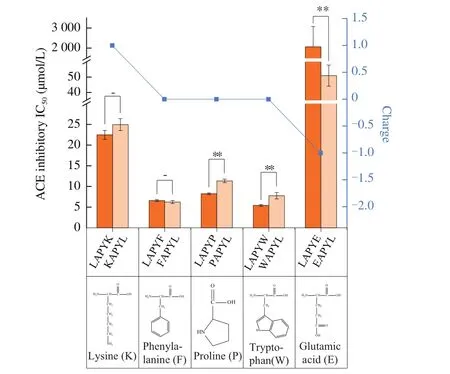
Fig. 2 The ACE inhibitory activities and the charges of modified peptides as well as the structures of their N/C-terminal amino acids. ** Represents that there was a significant difference (P < 0.01) between the ACE inhibitory activities of the peptides. The bar charts represent the ACE inhibitory IC50 of the peptides. The line in blue represents the charge of the peptides.
Fig. 2 illustrates the relationships between the hydrophobicity and the ACE inhibitory activities of peptides. The LAPYW (IC50of(5.42 ± 0.21) μmol/L) and the LAPYF (IC50of (6.58 ± 0.22) μmol/L)showed higher ACE inhibitory activities than others. Since the amino acids W and F were more hydrophobic than the C-terminal amino acids (P/K/E) of other three peptides, we could infer that containing hydrophobic amino acids was indeed beneficial for the ACE inhibitory activities of the peptides. Exchanging the N-terminal and the C-terminal amino acids of the LAPYE, we observed a significantly higher (P< 0.01) ACE inhibitory activity of the EAPYL (IC50of (50.97 ± 6.90) μmol/L) than that of the LAPYE.We inferred that the C-terminal hydrophobic amino acids exerted greater benefits to the ACE inhibitory activities of peptides than did the N-terminal hydrophobic amino acids.
Neither the order of the C-terminal amino acids nor the order of the peptides on the basis of hydrophobicity was consistent with the order of the peptides on the basis of ACE inhibitory activity. Thus, we investigated the plausible contributions of the amino acid structures towards the ACE inhibitory activities of the LAPYK and the modified peptides. We found that the side chains of the amino acids with ring structures (W, F, P) increased the ACE inhibitory activities of the peptides. Similar to our study, the study by Mirzaei et al. [35] reported the roles of aromatic amino acids in the ACE inhibitory activities of peptides. Besides, the potent ACEIPs IPP, VPP with proline ring at C-terminus also supported the observations [36].
3.3 The molecular-interaction mechanism between the peptides and ACE
Molecular docking was used to explore the interactions between the peptides and ACE. Two hundred poses of peptides were clustered using MGL Tools software [37]. Considering both the binding energy and poses cluster, the pose with the highest cluster aggregation and the lowest binding energy was extracted and used to analyze.Table 1 shows the intermolecular energy and the decomposition energy of the LAPYK and the modified peptides. The LAPYK and the LAPYE with C-terminal charged amino acid exhibited low electrostatic energies—the low energies favored the peptide bindings to ACE. However, as shown in Fig. 3, the Lys5 of the LAPYK and the Glu5 of the LAPYE were 12.08 and 9.28 Å apart from the zinc ion, respectively. Typically, the distance of the atoms which could form interactions was less than 3.5 Å. Thus, the Lys5 of the LAPYK and the Glu5 of the LAPYE could not lead to interactions with zinc,which was the active site of ACE [38]. Thus, the electrostatic energies of the LAPYK and the LAPYE were not inversely proportional to the ACE inhibitory activities of the peptides.
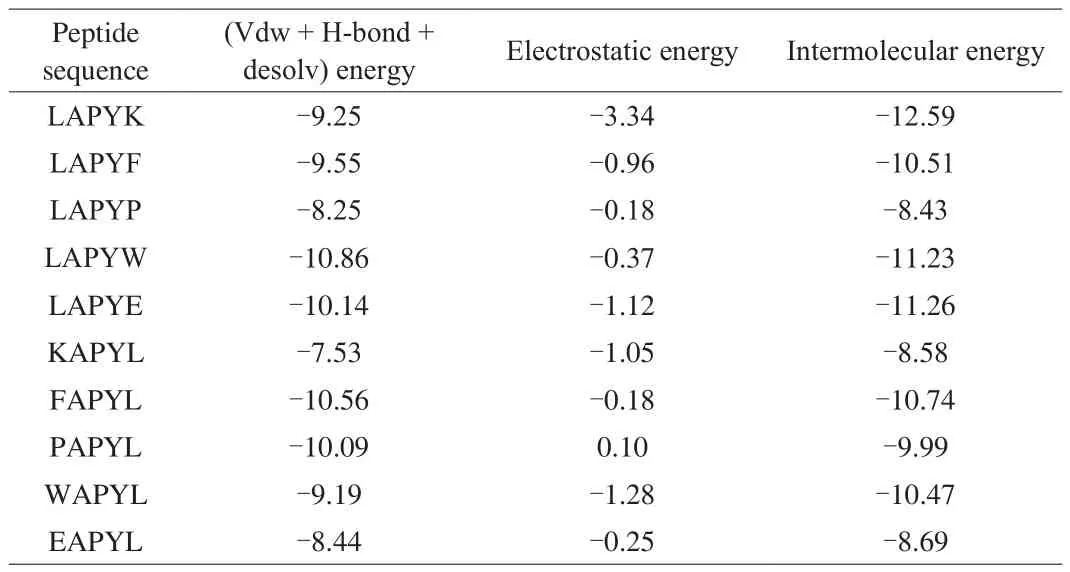
Table 1Intermolecular decomposition energy (kcal/mol) between peptides and ACE.
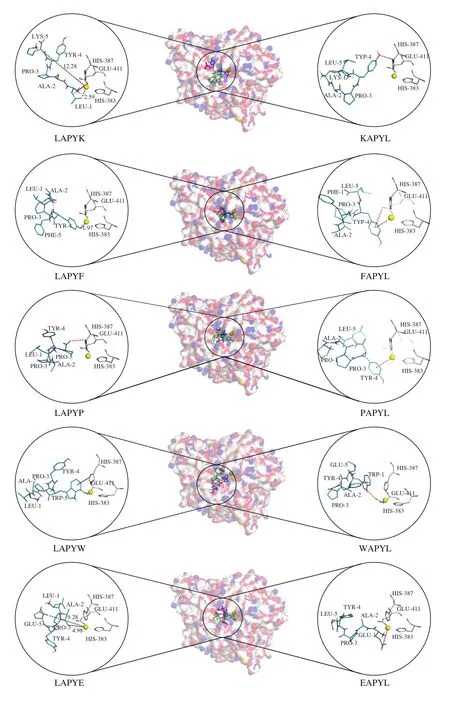
Fig. 3 The intermolecular interactions of peptides and ACE. The distances between atoms are indicated in grey, the hydrophobicity interactions are indicated in blue, the hydrogen bonds are indicated in orange, and the electrostatic interactions are indicated in red.
Several reports have indicated that ACEIPs have short distances to the zinc ion of ACE [39]. Similarly, our results indicated that the LAPYE that showed poor ACE inhibitory activity was located far from the zinc ion (4.98 Å). In contrast, the LAPYF with higher activity than LAPYE was located at close proximity to the zinc ion (1.97 Å). Although the Lys5 was far from the zinc ion, the Leu1 of LAPYK was nearer to the zinc ion (2.59 Å) than was the LAPYE.Thus, the LAPYK exhibited higher ACE inhibitory activity than did the LAPYE.
Except for the zinc ion, a HEXXH motif including His383, His387,and Glu411 were also reported to be the active site of ACE [40].The interactions between peptides and ACE were analyzed using Discovery Studio Visualizer 4.1 (DS Visualizer 4.1, Accelrys Software Inc. http://www.accelrys.com). As shown in Fig. 3, the Pro5 of LAPYP interacts with His387 via a hydrogen bond, whereas, the Trp5 of LAPYW forms hydrophobic interaction with His387. The interactions between peptides and ACE contributed to the high ACE inhibitory activities of the LAPYP and the LAPYW. Our findings were in agreement with the findings reported by Xu et al. [41].The authors identified an ACEIP, LVF, forms a hydrogen bond with His387. Similarly, Abdelhedi et al. [42] reported that the antihypertensive drug Lisinopril interacts with His387.
After the exchange of N/C-terminal amino acids, the LAPYF exhibited significant changes in neither the preferable docking pose nor the ACE inhibitory activity. It is worth noting that tyrosine (Y)plays an important role in the interactions between peptides and the ACE active sites. The Tyr4 of KAPYL formed a hydrogen bond with His387. On the other hand, the electrostatic interaction between Tyr4 and the zinc ion contributes a lot towards the inhibitory activities of the FAPYL and PAPYL. In addition, Tyr4 of FAPYL formed electrostatic interaction with Glu411 of ACE. Overall, thesefindings supported the benefits of the amino acids with ring structures to the ACE inhibitory activities of peptides.
3.4 The MD simulations of ACE-peptide complexes
MD simulations were performed for the ACE-LAPYK, the ACE-LAPYW, and the ACE-LAPYE complexes. The LAPYW and the LAPYE demonstrated the strongest and the weakest ACE inhibitory activities among the peptides, respectively. The root mean square deviation (RMSD) values of the backbone atoms and the radius of gyration (Rg) values were calculated as a measurement for the structural stability of ACE. The results are provided in Fig. 4. After 200 ns simulations, both the RMSD and the Rg values were stable,indicating that the peptides formed stable complexes with ACE.
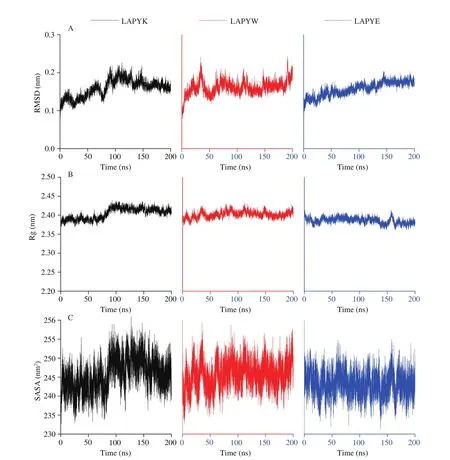
Fig. 4 The (A) RMSD, (B) Rg, (C) SASA values of ACE in the ACE-LAPYK, the ACE-LAPYW, and the ACE-LAPYE complexes.
In particular, the mean RMSD values of the ACE backbone were calculated for each ACE-peptide complex. The mean RMSD value for the ACE-LAPYW complex was 0.160 8, which was higher than those of the ACE-LAPYK (0.158 9) and ACE-LAPYK (0.156 8) complex.It meant that there were more conformational changes of ACE in the ACE-LAPYW complex than were there ACE conformational changes in the ACE-LAPYK and the ACE-LAPYE complexes.This observation justified the higher ACE inhibitory activity of the LAPYW than the ACE inhibitory activities of the LAPYK and the LAPYE. A previous study has shown the same finding that the peptide with higher activity possessed a higher RMSD of ACE [43].
Solvent accessible surface area (SASA) measures the protein surface area that is accessible for solvent and affects the ligand binding [44]. The SASA values of ACE in the ACE-LAPYK, the ACE-LAPYW, and the ACE-LAPYE complexes were calculated and are shown in Fig. 4. Among the SASA values of the complexes,the average SASA value of the ACE-LAPYW complex was the highest and that of the ACE-LAPYE complex was the lowest. The order of the complexes on the basis of average SASA value was in accordance with the order of the complexes on the basis of Rg value.The average SASA value of the ACE-LAPYW complex suggested that LAPYW binding to ACE loosened the ACE structure. As a result, the peptide could enter the cavity and interact with the active site of ACE more easily.
Root mean square fluctuations (RMSF) stands for the fluctuation of protein structure that is comparable to the X-ray B-factors [45].Low RMSF value represents a stable complex [46]. The RMSF values of ACE in the three ACE-peptide complexes were calculated and are shown in Fig. S1. From the results, we observed that RMSF of ACE in ACE-LAPYW complex was the lowest. It indicated that the peptide LAPYW could form a more stable complex with ACE,as a result, the peptide LAPYW was more likely to show ACE inhibitory activity.
3.5 MMPBSA Calculations
Twenty-nanosecond stable frames of complexes were extracted to calculate the binding free energy of the ACE-peptide complexes using MMPBSA method. The results were shown in Fig. 5, The binding free energy of the LAPYW (-64.66 kJ/mol) favored the LAPYW binding to ACE in comparison to the binding free energy of the LAPYK (-32.51 kJ/mol) and the LAPYE (4.13 kJ/mol). The binding energy reflects the strength of the interactions between ligand and protein [47]. The results of MMPBSA calculations predicted that the LAPYW demonstrated high ACE inhibitory activity and the LAPYE scarcely showed any ACE inhibitory activity; the predictions were in agreement with the results of ACE inhibitory activity investigations.
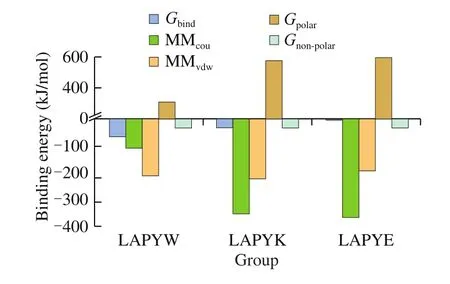
Fig. 5 The MMPBSA binding free energy between peptides and ACE.

Fig. 6 The ITC profiles of the 3 peptides (A: LAPYK, B: LAPYW, C: LAPYE) binding to ACE.
Comparing the charged peptides LAPYK and LAPYE with non-charged peptides LAPYW, we established that the ΔEcouvalues of the LAPYK(-367.80 kJ/mol) and LAPYE (-354.64 kJ/mol) were lower than that for the LAPYW (-109.37 kJ/mol). However, higher ΔGpolarvalues of the ACE-LAPYK complex (576.53 kJ/mol) and the ACE-LAPYE complex (598.62 kJ/mol) could be seen in comparison to the ACE-LAPYW complex (288.84 kJ/mol). Positive values meant that ΔGpolarwas unfavorable for the interactions between peptides and ACE [48]. Too higher positive ΔGpolarvalues explained why the LAPYK and LAPYE showed lower ACE inhibitory activity than did the LAPYW.
The ΔGnon-polarvalues of the three complexes were -32.31 kJ/mol(ACE-LAPYW), -31.47 kJ/mol (ACE-LAPYK), and -31.96 kJ/mol(ACE-LAPYE), respectively. The order of the complexes on the basis of ΔGnon-polarvalue was consistent with that on the basis of molecular weight. The peptide with higher molecular weight had a lower ΔGnon-polarvalue.The value of ΔGnon-polarwas negative, which was favorable for the interactions between the peptides and ACE. Thus,we could infer high molecular weight was beneficial for the ACE inhibitory activities of peptides.
3.6 The ITC analyses of the bindings between peptides and ACE
The thermodynamic profiles of the interactions between peptides and ACE were evaluated by ITC and are shown in Fig. 6. The association constant (Ka), the enthalpy change (ΔH), and the entropy change (ΔS) values were obtained after fitting of integrated heats.As shown in Fig. 6, the Ka((2.48 × 1011± 1.13 × 1011) L/mol) of the LAPYW was higher than that of the LAPYK ((2.55 × 106±1.32 × 106) L/mol), which was consistent with the higher ΔGbindingvalue of the LAPYW than that of the LAPYK. Lan et al. [49] reported that the Kaof the ACEIP GMKCAF with an IC50of 45.7 μmol/L was 9.78 × 103L/mol, which was lower than the Kavalues of the LAPYW and the LAPYK. It suggested that the Kacould be used to predict the ACE inhibitory activity of the peptides. A higher Karepresented a higher activity.
Different from the results of MD simulations, the results of ITC analyses suggested that the binding of LAPYE to ACE was favorable: the Kaof the peptide was (1.22 × 106± 5.50 × 105) L/mol.The deviation can be explained partly by ignoring the entropy term of the MMPBSA calculation. The ΔS value of the LAPYE was positive,which indicated that the reaction between the LAPYE and ACE was driven by the entropy change. Conversely, the ACE-LAPYW and the ACE-LAPYK interactions were driven by the enthalpy changes.Due to the higher ACE inhibitory activities of the LAPYW and the LAPYK than that of the LAPYE, we inferred that the enthalpy-driven interactions were more favorable than the entropy-driven interactions for facilitating the ACE inhibitory activities of peptides.
Although the ΔH values of the LAPYW and LAPYK were favorable for the peptides binding to ACE, the ΔS values of the peptides exert the opposite effects. This phenomenon of enthalpy-entropy compensation weakened the bindings between ACE and the peptides,thus, reducing the ACE inhibitory activities of the LAPYW and the LAPYK [50]. Wu et al. [51] measured the thermodynamics parameters of the interactions between ACE and the antihypertensive drug, Lisinopril.The results showed that the reaction of ACE-Lisinopril is driven by both the enthalpy and the entropy change. Therefore, we suggested that investigations for the peptides whose reaction with ACE are driven by both the enthalpy and the entropy change.
4. Conclusion
The structural requirements and intermolecular-interaction mechanism of ACEIPs were investigated in this study. Our results showed that the hydrophobicity and the amino acids with ring structures were beneficial for the ACE inhibitory activities of peptides.Based on the structural requirements evaluations, an ACEIP, LAPYW,was obtained; this peptide exhibited an IC50of (5.42 ± 0.21) μmol/L.The LAPYW displayed 4 times as high as ACE inhibitory activity than did the template peptide LAPYK. The polar solvation free energies limited the ACE inhibitory activities of the peptides LAPYW and LAPYE. In addition, although the enthalpy-driven reaction was more favorable than the entropy-driven reaction to exhibit ACE inhibitory activities for peptides, the phenomenon of enthalpy and entropy compensation might limit the ACE inhibitory activities of our peptides. Since the reaction between Lisinopril and ACE was driven by both enthalpy and entropy, we conjecture that the peptides which could bind to ACE driven by both the enthalpy and the entropy change might exhibit higher ACE inhibitory activities.Validation experiments should be performed in the future.
Declaration of con flicting interest
The authors declare no con flicting interest.
Acknowledgements
This work was funded by the National Natural Science Foundation of China (31972096), Jilin Province Science and Technology Youth Talent Support Project (QT202021) and Interdisciplinary Integration and Innovation Project of JLU (JLUXKJC2021QZ11).
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at http://doi.org/10.1016/j.fshw.2022.06.021.

- 食品科学与人类健康(英文)的其它文章
- A review on mechanisms of action of bioactive peptides against glucose intolerance and insulin resistance
- Adropin as an indicator of T2DM and its complications
- Isolation and characterization of novel peptides from fermented products of Lactobacillus for ulcerative colitis prevention and treatment
- The immunity-promoting activity of porcine placenta in mice as an immunomodulator for functional foods
- Nutritional properties of Europen eel (Anguilla anguilla) bone peptide-calcium and its apoptosis effect on Caco-2 cells
- A comprehensive method to explore inhibitory kinetics and mechanisms of an anticoagulant peptide derived from Crassostrea gigas