
高效液相色谱-串联质谱法检测泮托拉唑钠原料药中的水合肼
2022-07-06 14:27:37赵会明张振洋樊华军
上海医药 2022年11期
赵会明 张振洋 樊华军
摘 要 建立了泮托拉唑钠原料药中的基因毒性杂质水合肼的高效液相色谱-串联质谱(LC-MSMS)检测方法。采用反相色谱,以水-乙腈(含0.1%甲酸)为流动相,梯度洗脱,流速0.5 mL/min,以ESI正离子多反应监测(MRM)模式进行质谱检测。结果显示,水合肼的检测限和定量限可达到0.23、0.47 ng/mL,其在0.47~9.37 ng/mL浓度范围内线性关系良好(r=0.999 9),准确度试验中低、中、高浓度回收率均在81.6%~90.9%之间。在3批次泮托拉唑钠原料药中均未检出水合肼。
关键词 高效液相色谱-串联质谱法 基因毒性杂质 泮托拉唑钠 水合肼 痕量检测
中图分类号:R917; O657 文献标志码:A 文章编号:1006-1533(2022)11-0072-04
引用本文 赵会明, 张振洋, 樊华军. 高效液相色谱-串联质谱法检测泮托拉唑鈉原料药中的水合肼[J]. 上海医药, 2022, 43(11): 72-75.
Determination of hydrazine hydrate in pantoprazole sodium by high performance liquid chromatography-tandem mass spectrometry
ZHAO Huiming, ZHANG Zhenyang, FAN Huajun
[ICAS Testing Technology Service (Shanghai) CO., LTD., Shanghai 201100, China]
ABSTRACT To establish a high-performance liquid chromatography-tandem mass spectrometry (LC-MSMS) method for the determination of hydrazine hydrate in active pharmaceutical ingredient ( API) pantoprazole sodium. HPLC was carried out by reverse chromatography using water-acetonitrile containing 0.1% formic acid as flow phase and gradient elution at a flow rate of 0.5 mL/min. Mass spectrometry was performed with multi-reaction monitoring (MRM) in positive ESI mode. The detection and quantitative limits of hydrazine hydrate reached 0.23, 0.47 ng/mL and hydrazine hydrate showed good linear relationship in the range of 0.47-9.37 ng/mL (r=0.999 9). The recoveries of samples at low, medium and high-level concentrations reached 81.6% to 90.9% in the accuracy experiment. No hydrazine hydrate was detected in 3 batches of pantoprazole sodium.
KEY WORDS HPLC-tandem mass spectrometry; genotoxic impurities; pantoprazole sodium; hydrazine hydrate; trace determination
上消化道出血是近年的临床疾病中常见且多发的一种疾病,其临床表现为呕血、黑便等,如得不到及时有效治疗,可能引发失血性休克。泮托拉唑钠对于十二指肠溃疡、急性胃黏膜病变、复合性胃溃疡等急性上消化道出血的疗效较好[1]。泮托拉唑钠的合成工艺路线较长,其重要起始物料5-二氟甲氧基-2-巯基-1H-苯并咪唑的合成中使用的水合肼属于1类基因毒性杂质[2-5]。因为其对药物安全性的威胁,近年来监管机构对于药物中的基因毒性杂质的研究越来越重视。EMA(欧洲药品管理局)、ICH(人用药品技术要求国际协调理事会)先后发布了相关的指导原则,推荐使用毒理学关注阈值TTC 1.5 mg/d作为基因毒性杂质的可接受限度[6-7]。
水合肼的还原性极强,是药物合成中经常被当作还原剂使用,是已知的致癌物。对于水合肼的检测方法的报道,国内外都有过相关研究,包括GC、HPLC以及离子色谱等方法[8-11],然而这些方法的灵敏度和抗干扰能力在原料药和制剂的检测中受到了挑战。本研究报道了一个简单的衍生化的方法,使用LC-MSMS对泮托拉唑钠原料药中的水合肼进行检测。
1 材料和方法
1.1 仪器
5500三重四极杆质谱仪和1.7 LC-MSMS软件工作站(AB Sciex公司);Nexera UHPLC LC-30A(岛津公司);SQP万分之一分析天平(Sartorius公司);Poroshell 120 PFP液相色谱柱(3.0 mm×150 mm,2.7 mm;Agilent公司)。
1.2 试药
水合肼对照品(含量97.9 %,批号SHBK4829,Sigma-Aldrich公司);苯甲醛(分析纯,批号LG70U41,百灵威公司);乙腈(色谱纯,批号JA104330,默克公司);甲酸(色谱纯,批号C12122682,麦克林公司);苯甲酸(分析纯,批号20190430,国药化学试剂有限公司);泮托拉唑钠原料药供试品(市售,批号AK20-255798-1,AK20-255798-2,AK20-255798-3)。
1.3 溶液制备
1)溶剂 乙腈-纯化水(70∶30)。
2)衍生化试剂 移取0.50 g苯甲酸和4.0 mL苯甲醛用溶剂溶解并定容至100 mL。
3)对照品储备溶液 精密称量水合肼对照品到容量瓶中,用超纯水稀释到1 000 ng/mL。
4)对照品溶液 取对照品储备溶液适量,加入2 mL衍生化试剂,用溶剂稀释并配置成0.23、0.47、2.34、4.69(100%限度点)、7.03、9.37 ng/mL一系列不同浓度对照品溶液,60 ℃水浴振荡衍生1 h,其中0.47 ng/mL作为定量限溶液(LOQ),0.23 ng/mL作为检测限溶液(LOD)。
5)供试品溶液 精密称量泮托拉唑钠供试品,加入2 mL衍生剂后用溶剂定容至0.5 mg/mL,60 ℃水浴震荡衍生1 h。
6)加標样品溶液(100%限度点为例) 精密称量泮托拉唑钠供试品适量,加入对照品溶液和2 mL衍生剂,待溶解后定容至0.5 mg/mL。
1.4 方法
泮托拉唑钠制剂每日最大服药量为160 mg,在ICH M7规定中,水合肼口服每日可接受摄入量为39 mg/d,按照最严格的TTC法计算水合肼在活性医药成分(active pharmaceutical ingredient,API)中的残留限度不得超过9.375 mg/g。水合肼分子质量只有32,其活性高、色谱行为不佳和可检测性差的特点对于其痕量残留的直接定量难度很大。苯甲醛在弱酸酸性加热条件下能与水合肼发生衍生化反应,生成二苄肼(图1),其在液质联用分析中的响应很好。
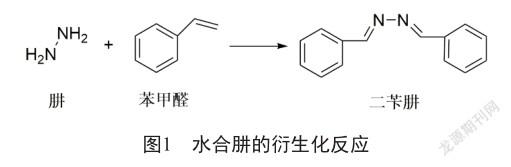
1.4.1 色谱条件
以0.1%甲酸水溶液为流动相A,0.1%甲酸乙腈溶液为流动相B,梯度洗脱(0~5 min,50%→75% B;5~8 min,75% B;8~9 min,75%→50% B;9~10 min,50% B),流速0.3 mg/mL,柱温40 ℃,进样体积2 mL,样品盘温度5 ℃。
1.4.2 质谱条件
采用电喷雾离子源(ESI),正离子多反应监测模式(MRM),设定气帘气压力35 psi,离子源温度600 ℃,离子化电压5 500 V,喷雾器压力50 psi,辅助加热气压力50 psi,水合肼的衍生化产物二苄肼的监测离子对信号为209.0 m/z(母离子)→109.0 m/z(子离子),DP电压80 V,CE电压24 V,信号采集时间5.0~7.5 min。
2 结果
2.1 专属性实验
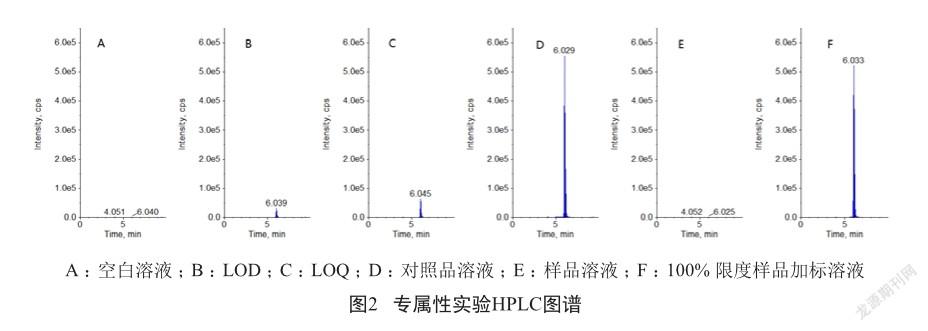
分别进样空白溶液,定量限溶液,对照品溶液,样品溶液,加标样品溶液,并记录色谱图(图2)。空白溶液色谱图中,目标峰处无干扰;对照品溶液色谱图中,6.04 min处显示二苄肼目标峰;100%限度样品加标溶液色谱图中,目标峰附近没有干扰峰。
2.2 线性关系考察
按照1.3中对照品溶液配置方式制成10%~200%限度溶液,相当于供试品0.94~18.75 mg/g浓度的一系列线性对照品溶液,进样测定。以浓度X (ng/mL)为横坐标,峰面积Y为纵坐标,线性回归方程为Y=837 631X-8 865,r=0.999 9。水合肼在0.47~9.37 ng/mL浓度范围内线性关系良好,Y轴截距占100%(4.69 ng/mL)响应值的百分比为0.2%。
2.3 定量限和检测限实验
取10%限度浓度为定量限浓度点(0.47 ng/mL)样品进样分析6次,其信噪比(S/N)分别为30,32,33,30,36,46,均大于10;6次定量限溶液峰面积的相对标准偏差RSD为0.6%;定量限点的加标样品溶液回收率为86.5%。
取5%限度浓度为检测限浓度点(0.23 ng/mL)样品进样分析3次,其信噪比(S/N)分别为15,18,15,均大于3,表明灵敏度良好。
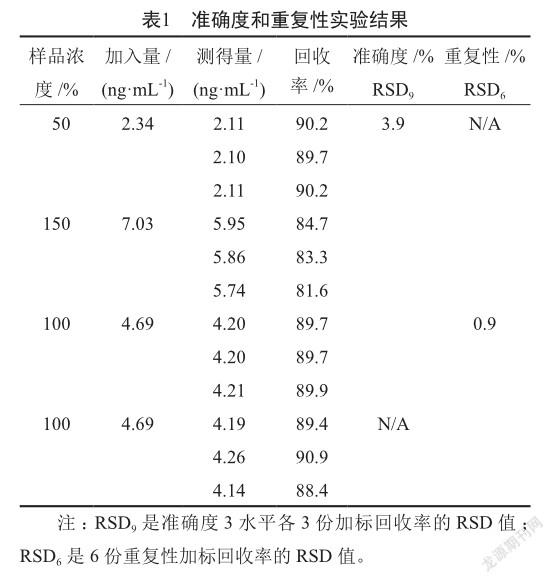
2.4 准确度和重复性实验
配制50%、100%和150%三个限度水平的样品加标溶液各三份,作为准确度测试溶液,每份溶液进样分析一次,每个浓度水平的加标回收率和9份回收率的RSD结果见表1。配制6份100%限度水平的加标样品溶液作为重复性溶液,每份溶液进样分析一次,6份回收率的RSD结果见表1。这些结果符合中国药典9101分析方法验证指导原则中的相关要求,表明该方法可靠。
2.5 溶液稳定性实验
分别配制100%限度水平的对照品溶液和加标样品溶液各一份,在0~24 h进样分析。各时间点峰面积与0 h的比值均在88%~96%之间,表明对照品溶液和加标样品溶液在24 h内稳定。
2.6 耐用性实验
取100%限度的加标样品溶液,分别改变柱温(±2℃),起始有机相比例(±2%)和流速(±0.02 mg/mL),每改变一个条件测定2次。耐用性条件下加标样品溶液回收率在81.5%~ 94.5%之间,色谱条件的小幅度变化不影响水合肼的定量检测,表明该方法耐用性良好。
2.7 供试品测定
按“1.3”配制3批次供试品溶液,每个批次配样两份进行测定,3批次泮托拉唑钠供试品中在检出限(0.94 mg/g)水平均未检出水合肼(图2 E)。
3 讨论
3.1 基质干扰的排除
由于泮托拉唑鈉的样品峰和二苄肼的目标峰保留时间相近,为了避免痕量检测中的基质效应对于方法回收率的影响,分别试用了Waters ACQUITY UPLC HSS T3色谱柱(3 mm×100 mm,1.8 mm)、Agilent Poroshell 120 EC-C18色谱柱(4.6 mm×150 mm,2.7 mm)、Agilent Poroshell 120 PFP色谱柱(3.0 mm×150 mm,2.7 mm),前两款色谱柱目标峰与样品峰不能很好分离,与原料药共流出使回收率偏低,更换Agilent Poroshell 120 PFP 色谱柱后能将目标峰和API主峰实现很好的分离,且只将5.0~8.0 min时间段接质谱检测器,其他时间段均切进废液,极大地降低了基质干扰。
3.2 衍生化条件的优化
分别考察了于室温和40、60、80 ℃温度条件下、10 min、30 min、1 h、2 h进行水合肼衍生化反应,结果发现,较高温度下反应速率较快。综合评估后将最佳反应温度和时间定为60 ℃、1 h,此条件下衍生化产物二苄肼能稳定存在。
本实验建立了一个肼衍生化—LC-MSMS方法,用于测定泮托拉唑钠中的基因毒性杂质水合肼,其与已报道的分光光度法和气相色谱法相比[12-14],灵敏度更高,专属性更强,但是LC-MSMS的仪器设备较为昂贵,不利于普遍使用。该衍生化方法简单,易于操作,且灵敏度高(检测限可达0.23 ng/mL),适用于药物中水合肼的检测。
参考文献
[1] 王文杰. 消化性溃疡合并上消化道出血应用泮托拉唑治疗的疗效观察[J]. 中国社区医师, 2020, 36(12): 28-29.
[2] 李菁, 李荣东, 刘磊. 左旋泮托拉唑钠、镁的合成[J]. 广州化工, 2016, 44(6): 58-59; 103.
[3] Leakakos T, Shank RC. Hydrazine genotoxicity in the neonatal rat[J]. Toxicol Appl Pharmacol, 1994, 126(2): 295-300.
[4] Hazardous Substance Database (HSDB): Hydrazine (302-01-2)[EB/OL]. (2005-06-24) [2021-06-22]. https://pubchem.ncbi. nlm.nih.gov/compound/9321.
[5] Carcinogenicity Potency Database (CPDB) [EB/OL]. [2021-07-03]. https://files.toxplanet.com/cpdb/cpdb.html.
[6] Guideline on The Limits of Genotoxic Impurities, 2006 [EBOL]. (2006-06-28)[2021-07-03]. https://www.ema.europa.eu/ en/documents/scientific-guideline/guideline-limits-genotoxicimpurities_en.pdf.
[7] ICH M7(R1) Assessment and Control of DNA Reactive(Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk, 2017 [EB/OL]. [2021-07-03]. https://www. ema.europa.eu/en/ich-m7-assessment-control-dna-reactivemutagenic-impurities-pharmaceuticals-limit-potential.
[8] 王汝金, 朱建德, 张丽芳, 等. 高效液相色谱法测定沙咪珠利中的水合肼残留量[J]. 中国兽药杂志, 2017, 51(11): 59-64.
[9] Elder DP, Snodin D, Teasdale A. Control and analysis of
hydrazine, hydrazides and hydrazones—Genotoxic impurities in active pharmaceutical ingredients (APIs) and drug products[J]. J Pharm Biomed Anal, 2011, 54(5): 900-910.
[10] 向彩红, 叶英, 姚劲挺, 等. 顶空-GCMS法测定药物中水合肼含量[C]//第六届全国药物分析大会论文集. 北京: 全国药物分析大会理事会, 2016: 1-2.
[11] 黄伟民, 黄伟静, 陈伟翰. GC-MS法测定苯磺酸氨氯地平原料药中水合肼的含量[J]. 中国药师, 2021, 24(1): 202-205.
[12] 胡平, 赵世民. 糠醛衍生化-气相色谱(NPD)法测定水中水合肼[J]. 地球与环境, 2017, 45(2): 242-246.
[13] 周延生, 李赛钰, 韩东升, 等. 药物中水合肼残留量的气相色谱分析[J]. 分析试验室, 2008, 27(3): 84-86.
[14] 郑筠. 对二甲氨基苯甲醛分光光度法测定水中水合肼[J].供水技术, 2017, 11(4): 50-52.
猜你喜欢
氯碱工业(2022年6期)2022-11-21 01:41:32
化工环保(2021年2期)2021-04-25 13:42:34
云南化工(2020年5期)2020-06-12 09:26:40
盐科学与化工(2019年11期)2019-12-04 02:16:42
分析化学(2017年1期)2017-02-06 21:34:35
中国纤检(2016年12期)2017-01-20 09:28:19
热带农业科学(2016年10期)2016-12-12 01:52:56
分析化学(2016年7期)2016-12-08 00:57:07
中国科技博览(2016年18期)2016-10-19 11:09:28
科学与财富(2016年28期)2016-10-14 04:01:52
