
细胞焦亡的研究进展及多糖在细胞焦亡中的潜在作用
2022-07-02 03:49:38范文涛吴陈颢金周雨
食品科学 2022年11期
范文涛,吕 凯,吴陈颢,金周雨,姜 雨,*,宋 慧,2,*
(1.吉林农业大学生命科学学院,吉林 长春 130118;2.吉林农业大学 教育部食药用菌工程研究中心,吉林 长春 130118)
焦亡是一种以促炎形式调节细胞程序性死亡的方式,与凋亡、坏死以及铁死亡不同,其特点是依赖炎症胱天蛋白酶(Caspases),并伴有大量的促炎细胞因子白细胞介素(interleukin,IL)-1β和IL-18的释放。多种物质可以通过刺激模式识别受体,激活焦亡相关信号通路,产生细胞因子,促使细胞膨胀、爆裂,引发细胞焦亡。近年来研究表明,细胞焦亡存在于多种疾病的发生发展过程中,如感染性疾病、心脏病、动脉粥样硬化、癌症、消化道疾病、糖尿病等。因此,这种新型程序性死亡方式üü细胞焦亡受到学者们广泛关注。
多糖是一种由多种单糖通过糖苷键连接而成的碳水化合物聚合物,在生物体内具有重要的作用。据报道,多糖具有调节免疫、抗氧化、抗肿瘤等多种生物活性,且具有毒性低、副作用小等诸多优点。但多糖是否可以通过目前已被研究的相关信号通路调节细胞焦亡鲜见报道,本文通过查阅近年来国内外研究报道,试图阐明细胞焦亡机制、引起焦亡信号的通路与各类疾病的关系,以及多糖在细胞焦亡相关信号通路中的作用,探讨多糖对细胞焦亡的潜在影响。
1 细胞焦亡
“细胞焦亡”一词是由Boise等最先使用,采用希腊语“Pyro”用来表示细胞死亡过程中伴随炎症产生的特征,与凋亡(Apoptosis)类似,结合与词根“ptosis”组合而成,命名为焦亡。在侵袭性致病菌志贺氏菌和沙门氏菌感染巨噬细胞过程中,首次观察到细胞焦亡的发生,它们通过分泌的效应蛋白沙门氏菌侵袭蛋白B(invasion protein B,SipB)和细胞核因子(nuclear,factor,NF)-κB抑制蛋白(inhibitor of NF-κB,IκB)激活巨噬细胞表达Caspase-1,从而引发细胞死亡。起初,此死亡过程被归类为凋亡,但在后续研究中发现感染过程中Caspase-1引起的细胞死亡过程与传统的凋亡过程有很大差别。此死亡过程迅速,细胞膜的完整性被破坏,并伴随着明显的炎症反应发生,因此从凋亡中区分出来,而作为另一种程序性死亡方式。随着对细胞焦亡研究的不断深入,2018年细胞死亡命名委员会将细胞焦亡概念修正为:一种依赖于Gasdermin家族蛋白形成质膜膜孔的可调控的细胞死亡,并非完全依赖炎性Caspase的活化而完成。
1.1 细胞焦亡的特征
典型的细胞凋亡特征(内在和外在)表现为:细胞核固缩、核染色质边集、细胞膜及各细胞器膜完整、膜可发泡成芽形成凋亡小体与细胞内成分分离、不引起周围组织炎症反应和修复再生。因此细胞凋亡是免疫沉默的过程。焦亡不同于凋亡,但也具有相似的特征,如细胞核固缩、染色质DNA断裂等。细胞焦亡也具有其独特的形态学特征,包括细胞膜上形成10~15 nm的小孔、细胞内离子平衡被破坏、水内流、细胞肿胀继而破裂、细胞发生渗透性溶解并且引起周围组织炎症。
1.2 细胞焦亡的分子机制
1.2.1 细胞焦亡的经典途径
细胞内外存在多种模式识别受体,当细胞受到损伤相关模式分子模型(damage-associated molecular patterns,DAMP)或病原相关模式分子(pathogenassociated molecular patterns,PAMP)刺激时,便会促进炎症小体相关蛋白组装活化(炎症小体根据受体蛋白不同分为核苷酸结合寡聚化结构域样受体蛋白(NOD-like receptor protein,NLRP)1、NLRP2、NLRP3、黑素瘤缺乏因子2(absent in melanoma 2,AIM2)及核苷酸结合寡聚化结构域(nucleotide-binding oligomerzation domain,NOD)样受体C4(NOD-like receptor C4,NLRC4)等),被活化的炎症小体会进一步激活炎性Caspase蛋白,如Caspase-1,活化的Caspase-1通过剪切前体pro-IL-1β和pro-IL-18使其成为有活性的IL-1β、IL-18,活化的Caspase-1也可通过切割Gasdermin家族蛋白,如Gasdeimin D,形成活性N-末端p30片段。16个N-末端片段相互组合形成直经10~15 nm的闭环结构,并锚定到细胞膜上,促使细胞因子释放导致炎性细胞死亡。在Caspase-1依赖的焦亡经典信号通路中,炎症小体起着举足轻重的地位(图1)。
炎症小体被激活的过程可分为启动和激活两个阶段(图1)。启动阶段:由微生物分子或内源性细胞因子刺激,通过激活转录因子NF-κB导致NLRP3和pro-IL-1β上调。Caspase-8和FADD通过激活NF-κB途径参与启动。Lys63特异性去泛素化酶BRCC36和IRAK1独立于转录调控NLRP3炎症小体的活化。激活阶段:通过多种刺激物如ATP、成孔毒素、病毒RNA和颗粒物等刺激使NLRP3、ASC、Pro-Caspase-1及调节蛋白Nek7组装形成炎症小体。炎症小体组装后,Pro-Caspase-1通过诱导邻近的自身蛋白溶解被激活,形成有活性的Caspase-1,完成NLRP3炎症小体激活。NLRP3炎症小体可以被多种刺激激活,其中K流出是NLRP3炎症小体激活的充分和必要条件。Ca信号转导可能导致线粒体功能紊乱,并与NLRP3炎症小体活化有关。线粒体功能障碍衍生信号,如mtROS、mtDNA或心磷脂外化,也被认为可介导NLRP3的激活。MAVS可介导RNA病毒诱导NLRP3活化。溶酶体破裂引起的K流出和组织蛋白酶的释放激活NLRP3。

图1 细胞焦亡经典与非经典途径信号通路指示图[23,26-31]Fig.1 Classical and non-classical signaling pathways of pyroptosis[23,26-31]
作为细胞焦亡经典途径核心的炎症小体,其活化机制尚未获得统一认识。其中K外排在NLRP3激活中的作用已被证实,而Ca信号、活性氧(reactive oxygen species,ROS)和线粒体功能障碍的作用仍然没有统一的结论。因此,整合各类刺激条件在激活炎症小体中的作用是未来研究细胞焦亡需要解决的问题。
1.2.2 细胞焦亡非经典途径
细胞焦亡非经典途径是对革兰氏阴性菌的独特免疫反应。如图1所示,在非经典途径中,脂多糖(lipopolysaccharide,LPS)通过转染或感染进入胞浆,激活Caspase-4、5、11。激活的Caspase-4、5、11通过裂解消皮素D(gasdermin D,GSDMD)形成N端p30结构,进而通过在细胞膜上形成孔来破坏细胞功能,触发细胞焦亡。活化的Caspase-4、5、11也通过触发Pannexin-1通道的开放,促使K外排,进而诱导NLRP3炎症小体激活以及IL-1β释放。从Pannexin-1通道释放的ATP激活P2X7R,进一步促进K外排,刺激炎症小体的组装,引发细胞焦亡。在非经典的焦亡信号通路中,焦亡的过程不需要Caspase-1,但IL-1β和IL-18的分泌需要Caspase-1的介导。目前对细胞焦亡非经典途径的研究尚不全面,与焦亡经典途径之间的关系还需深入研究。
1.2.3 细胞焦亡的其他途径
除细胞焦亡经典途径与非经典途径外,还存在由Caspase-3、颗粒酶等诱导的细胞焦亡。因没有统一的归类方式,本文现将其归类为细胞焦亡其他途径(图2)。Zhou Zhiwei等发现细胞毒性淋巴细胞(如细胞毒性T淋巴细胞(cytotoxic T lymphocyte,CTLs)、自然杀伤细胞(natural killer cell,NK)等)中的颗粒酶A(granzyme A,GzmA)可以经穿孔素(perforin)进入靶细胞,通过水解消皮素B(gasdermin B,GSDMB)分子Lys229/Lys244位点诱导靶细胞发生焦亡(图2左)。有研究发现,传统诱导细胞凋亡的关键蛋白Caspase-3也可诱导细胞焦亡的发生。Caspase-3诱导的细胞焦亡与Caspase-1、4、5/11切割GSDMD诱导的细胞焦亡机制不同。Caspase-3切割Gasdermin家族中另一成员Gasdermin-E(GSDME)生成N-末端片段(GSDME-N),该片段通过在质膜上形成孔来执行焦亡。在嵌合抗原受体T细胞(chimeric antigen receptor T,CAR-T)免疫疗法中,CAR-T细胞释放的GzmB可以通过穿孔素进入细胞,并活化Caspase-3,活化的Caspase-3通过切割GSDME来激活Caspase非依赖性的焦亡(图2中)。在高表达GSDME的肿瘤细胞中,用传统的化疗药物处理细胞,可以使凋亡关键蛋白Caspase-3活化,识别内源性GSDME,导致细胞焦亡,进而控制肿瘤细胞生长(图2右)。由于细胞中存在转运必需内体分选复合物(endosomal sorting complex required for transport,ESCRT)介导的孔修复机制,因此GSDME的表达量决定着细胞是凋亡还是焦亡,通常在GSDME低表达的细胞中发生凋亡,而GSDME高或中等表达的细胞则直接发生GSDME介导的焦亡。

图2 细胞焦亡其他途径信号通路指示图[45,49-50]Fig.2 Other signaling pathways of pyroptosis[45,49-50]
细胞焦亡其他途径主要在肿瘤细胞中被发现,并且在肿瘤发生发展过程中占据重要位置,现已成为一种克服肿瘤耐药性、提高抗肿瘤药物治疗效果的一种新方法。
2 细胞焦亡与相关疾病的关系
细胞焦亡是一种促炎性细胞死亡,也是集促炎与细胞死亡于一身的新型细胞死亡方式,在多种疾病中发挥着重要作用。NLRP3等炎症小体活化及下游效应分子的激活存在于焦亡经典途径与非经典途径中,故探究NLRP3炎症小体及其下游效应分子的表达可间接了解细胞焦亡情况。在焦亡其他途径中Caspase-3在介导肿瘤杀伤过程中起到至关重要的作用。
2.1 焦亡经典途径与疾病的关系
NLRP3炎症小体作为焦亡经典途径的核心,在细胞焦亡中起着至关重要的作用。依赖NLRP3炎症小体的细胞焦亡导致的IL-1β水平增加和长时间局部炎症影响着免疫性疾病及肿瘤相关疾病。在阿尔茨海默症中,NLRP3炎症小体在小胶质细胞内聚集激活时,导致Caspase-1的裂解和下游IL-1β的活性增加。同时,NLRP3的激活会使Tau激酶和磷酸酶活性降低,进一步抑制Tau高磷酸化和聚集,并进一步促进阿尔茨海默症的病程进展。人类免疫缺陷病毒(human immunodeficiency virus,HIV)对小胶质细胞也具有相似的作用,HIV-1壳膜蛋白结构GP120可诱导小胶质细胞NLRP3依赖性焦亡和IL-1β的产生。抑制小胶质细胞NLRP3炎性体的激活,可以减轻GP120介导的神经炎性因子释放和神经元损伤。通过缓慢给药方式向转基因小鼠注射新型选择性NLRP3抑制剂MCC950后,不仅可以减轻神经炎症和神经元死亡,而且还可以促进神经元再生,恢复受损的神经认知功能。在艾滋病诱发的肾病中,HIV会以剂量和时间依赖性的方式通过NLRP3/Caspase-1途径诱导足细胞的焦亡,加快艾滋病相关肾病的发展。除HIV病毒外,结核分枝杆菌侵入机体后可以通过Caspase-1/NLRP3/GSDMD介导人单核细胞和巨噬细胞的焦亡,此方式是结核分枝杆菌感染引起细胞毒性的主要方式。川崎病(Kawasaki disease,KD)是一种免疫性的血管炎,有研究发现,由高迁移率族蛋白B1((high-mobility group box 1 protein,HMGB1)/晚期糖基化终产物受体(receptor for advanced glycation endproducts,RAGE)/组织蛋白酶B(cathepsin B)信号激活的NLRP3炎症小体依赖的内皮细胞焦亡是导致KD冠状动脉内皮损伤和炎症反应的重要诱因。痛风性关节炎(gout arthritis,GA)是一种持续性高尿酸血症期间因尿酸盐沉积在关节而引起的常见炎症性关节炎,其发生常伴随着细胞焦亡。尿酸钠(monosodium urate,MSU)作为GA的主要诱导因子,可以通过溴结构域蛋白4(bromodomain-containing protein 4,BRD4)/NF-κB/NLRP3/GSDMD信号传导导致大鼠滑膜中巨噬细胞焦亡,BRD4特异性抑制剂(JQ-1)能够抑制这种细胞焦亡的发生。有研究发现,在由MSU诱导的GA大鼠和成纤维样滑膜细胞(fibroblast-like synoviocytes,FLS)模型中,MSU降低了FLS细胞的活力并促进FLS细胞的焦亡,上调了GA大鼠滑膜组织中NLRP3、ASC、裂解的Caspase-1、裂解的N端GSDM和IL-1β的表达。微小RNA223-3p(miR-223-3p)通过靶向NLRP3抑制MSU诱导的大鼠和FLS细胞的焦亡。
在肿瘤细胞的发生发展中,NLRP3炎症小体介导的细胞焦亡具有双重作用。一项研究发现,沥青颗粒(coal tar pitch extract,CTPE)或LPS联合CTPE处理人正常肺细胞(BEAS-2B),可以体外诱导NLRP3小体的激活并伴随IL-1β的释放。在裸鼠体内成瘤过程中发现,两种处理均能导致裸鼠体内形成炎症微环境,使正常细胞向恶性细胞转化。另一项体外实验也具有相似的现象,使用LPS联合ATP处理A549细胞可以激活NLRP3炎性小体和并诱导细胞因子的释放,形成炎症微环境,促进A549肺癌细胞的增殖和迁移。另外,在先天免疫在癌症免疫监测中,使用C57BL/6脾内注射小鼠原发性结肠癌细胞(MC38)的荷瘤小鼠模型中,NLRP3炎性小体的激活能够介导下游IL-18成熟与释放,促进NK细胞成熟增强其杀瘤活性,抑制结肠癌MC38细胞肝转移生长。相似的结果也存在于重楼皂苷VI(polyphyllin VI,PPVI)处理的A549和H1299细胞中,PPVI通过激活A549和H1299细胞中的ROS/NF-κB/NLRP3/GSDMD信号轴,诱导Caspase-1介导的焦亡性细胞死亡。
NLRP3依赖的细胞焦亡在各类疾病中发挥着不同的作用,NLRP3炎症小体活化与细胞焦亡发生之间的关系仍然不明确,以及NLRP3炎症小体激活引发各类疾病是否均引起细胞焦亡仍不明朗。需要进一步的研究来阐明在各类疾病中NLRP3炎症小体活化与细胞焦亡的具体机制,以及在疾病中所起的作用。
2.2 焦亡非经典途径与疾病的关系
焦亡非经典途径主要由胞内LPS感受器Caspase-4/5/11与革兰氏阴性细菌的相互作用所触发。因此非经典焦亡多发生在由细菌感染引发的疾病中。来自革兰氏阴性细菌(包括大肠杆菌、鼠伤寒沙门氏菌、志贺氏菌)的LPS会激活小鼠Caspase-11,导致细胞焦亡,引发致死性败血性休克。在此过程中,HMGB1发挥着至关重要的作用,在脓毒血症发生的过程中,肝细胞释放的HMGB1将体内循环中的LPS转运至血管内皮细胞和巨噬细胞的胞浆内,并启动Caspase-11介导的细胞焦亡,进而引发败血症的发生。脓毒血症是各种临床急危重疾病常见并发症之一,严重时可引起多器官功能障碍。已有研究发现在脓毒血症模型小鼠中,会存在Caspase-11/GSDMD途径触发的细胞焦亡并介导铁蛋白的分泌,引起强烈的感染性损伤。除细菌感染外,非经典焦亡途径也存在于过敏性气道炎中,有研究发现,Caspase-4/11参与过敏性气道炎的发生发展,并且前列腺素E2(prostaglandin E2,PGE2)可以负调控由Caspase-11介导的细胞焦亡。
2.3 焦亡其他途径与疾病的关系
焦亡其他途径中的信号转导途径均在肿瘤细胞中发现,因此,针对细胞焦亡其他途径,本文重点探讨其与肿瘤的关系,Caspase-3及免疫细胞释放的颗粒酶是该途径的关键分子,其中Caspase-3依赖的细胞焦亡在焦亡其他途径中发挥重要的作用。Caspase-3依赖的细胞焦亡途径作为抗肿瘤耐药性的热点途径已经备受关注。有研究发现,冷空气等离子体可以通过刺激肿瘤细胞产生ROS诱导Caspase-3/GSDME依赖性的细胞焦亡信号通路,进而抑制肿瘤的生长。二甲双胍可作为增敏剂放大AMP活化蛋白激酶(AMP-activated protein kinase,AMPK)/凋亡相关蛋白沉默信息调节子1(silent informa tion regulator protein 1,SIRT1)/NF-κB信号,造成线粒体功能紊乱,ROS水平升高,诱导Caspase-3/GSDME介导的癌细胞焦亡。化疗药物洛巴铂、紫杉醇和顺铂均可以介导ROS/Jun氨基末端激酶(Jun N-terminal kinase,JNK)/Bax线粒体凋亡途径,Caspase-3/GSDME依赖性的细胞焦亡对癌症起到治疗作用,顺铂比紫杉醇更能诱导A549细胞焦亡。在黑色素瘤细胞中,铁可以显著增加化疗引发的ROS水平,促进线粒体外膜蛋白Tom20的氧化和寡聚化,活化的Tom20招募Bax到线粒体,诱导细胞色素c的分泌,激活Caspase-3,最终裂解GSDME诱导黑色素瘤细胞焦亡。GSDME表达水平调控凋亡-焦亡之间的转换,在肺癌、胃癌和黑色素瘤等多种肿瘤治疗过程中发挥重要作用。细胞焦亡在抗肿瘤中发挥的作用仍不清楚,何种条件能够引起肿瘤细胞的焦亡仍在积极的探索中,但随着对细胞焦亡研究的不断深入,相信会有更加明确的机制被阐述。
3 多糖在细胞焦亡中的潜在作用
细胞焦亡受多种因素直接或间接调控,存在多条途径,其中NF-κB/NLRP3/Caspase-1/GSDMD信号轴是细胞焦亡最经典的途径,ROS/Caspase-3/GSDME信号轴是肿瘤细胞焦亡的主要信号通路。已证实多糖可调节机体免疫、抗氧化以及诱导细胞的自噬与凋亡,在抗肿瘤中起到直接或间接的作用。多糖对细胞焦亡的作用仍没有详细、全面报道,现多集中在对焦亡信号通路中关键靶蛋白的调节作用,如对NF-κB、NLRP3炎症小体及细胞中ROS的调节作用。
3.1 多糖对细胞焦亡经典途径的作用
如前所述,细胞焦亡经典通路的激活主要分为两个阶段:启动阶段和激活阶段。在启动阶段涉及NF-κB信号通路激活,激活阶段主要是炎症小体的活化以及下游蛋白的活化。而许多研究发现,多糖分别对NF-κB及NLRP3炎症小体具有调节作用。有关多糖影响焦亡经典途径相关文献总结见表1。
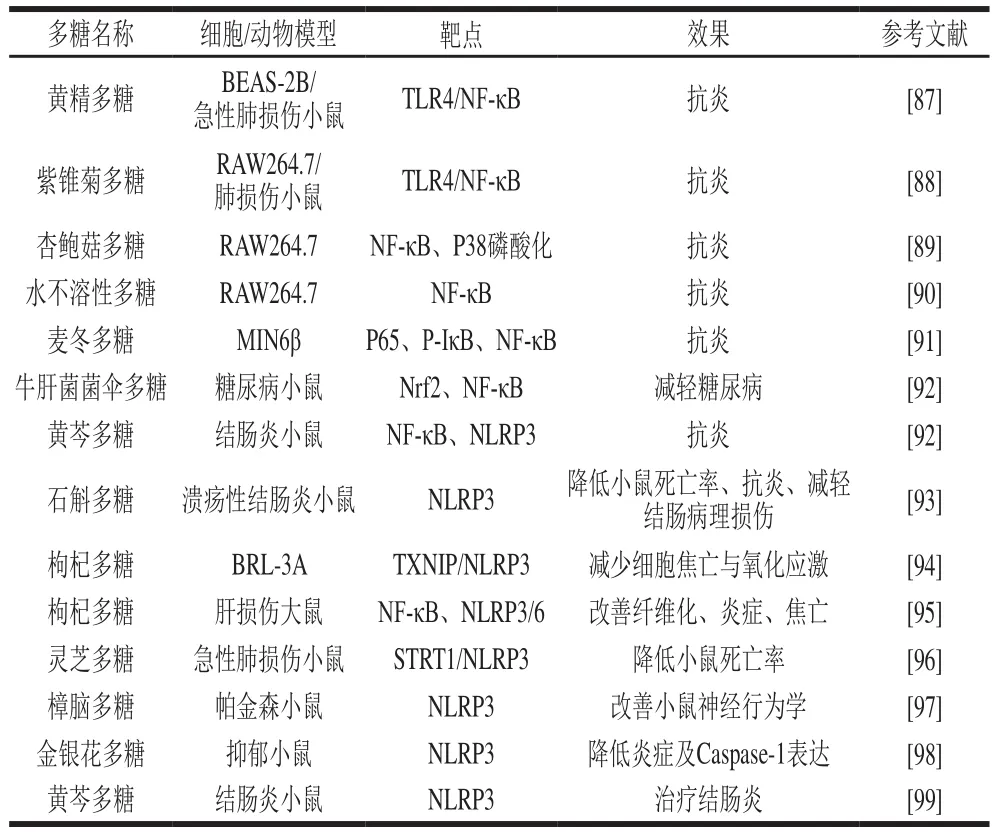
表1 多糖影响焦亡经典途径关键靶点及其调控效果Table 1 Polysaccharide affect the key targets of the classical signaling pathway of pyroptosis and their regulatory effects
3.1.1 多糖对细胞经典焦亡途径启动阶段的调节
细胞焦亡经典途径的激活主要分为启动阶段与激活阶段,其中启动阶段是为激活阶段准备所需要的“物质条件”。在启动阶段NF-κB信号通路起着至关重要。NF-κB信号通路的作用已被证明与促炎细胞因子生成有关。已有研究表明多糖可以通过调节NF-κB信号通路调节细胞的免疫应答。黄精多糖可以减轻LPS诱导的急性肺损伤大鼠肺病理变化,抑制了LPS诱导的中性粒细胞比例增加,并降低支气管肺泡灌洗液中的炎症因子水平。此外,在体外,黄精多糖降低了LPS诱导的炎症因子水平增加,黄精多糖还通过抑制TLR4/NF-κB途径抑制炎症,从而减少了LPS诱导的BEAS-2B细胞死亡。紫锥菊多糖与黄精多糖作用类似,也可通过抑制炎症反应和细胞死亡来减轻LPS诱导的小鼠肺损伤,并且抑制LPS诱导的RAW264.7细胞炎症因子表达,作用与TLR4/NF-κB信号通路有关。两种杏鲍菇多糖(WPEP、NPEP)在LPS刺激的RAW264.7巨噬细胞中表现出良好的抗炎作用。WPEP和NPEP可通过调节NO、PGE2、IL-1β、TNF-α和IL-6的产生来显著抑制LPS诱导的炎症反应。两种多糖组分抑制RAW 264.7巨噬细胞p38磷酸化,通过调节NF-κB相关的DNA表达来调节炎症因子的分泌。相似的结论也出现在不易消化的水溶性多糖处理的LPS诱导的RAW264.7细胞炎症模型中,NF-κB的活化也被显著抑制,从而降低了巨噬细胞的炎症反应。麦冬多糖能够通过抑制NF-κB途径中p65和p-IκB活化,增强葡萄糖刺激胰岛素分泌效应,降低了MIN6 细胞中IL-1β的分泌。牛肝菌菌伞中提取的多糖(SLPC-1S)可以通过调节Nrf2介导的氧化应激和NF-κB介导的炎症反应而预防和治疗链球菌素诱发的糖尿病。有研究发现,黄芩多糖(SP1-1)能够改善葡聚糖硫酸钠(dextran sulfate sodium,DSS)诱导的结肠病理损伤,并降低髓过氧化物酶活性。SP1-1能明显抑制DSS诱导的结肠炎小鼠血清和结肠中的促炎细胞因子水平,包括IL-1β、IL-18和TNF-α。此外,在SP1-1饲喂的小鼠结肠中检测到CD11b巨噬细胞浸润的减少和腹膜巨噬细胞中Caspase-1的失活。SP1-1具有这些作用的机制归因于其对NF-κB信号传导和NLRP3炎性小体激活的抑制。多糖可以通过调控NF-κB信号通路减轻机体或细胞中的炎症反应,但多糖调节细胞焦亡及炎症反应的研究较少。
3.1.2 多糖对细胞经典焦亡途径激活阶段的调节
炎性小体是一种蛋白复合物,在多种炎症疾病中起重要作用。炎性小体是先天免疫系统的一部分,可触发炎症细胞因子IL-1、IL-18的激活。炎性小体处于经典焦亡途径激活阶段的核心地位,对于经典焦亡途径的激活起着至关重要的作用。在众多炎症小体中,NLRP3炎性小体是的研究是最多也是最透彻的。很多研究显示多糖能够通过调节NLRP3炎症小体活化改善机体炎症的发生。石斛多糖可以明显抑制溃疡性结肠炎(ulcerative colitis,UC)小鼠中NLRP3炎症小体的激活,改善DSS诱导的急性UC小鼠的临床症状和体征,降低死亡率,减轻结肠病理损伤,重建炎症因子和抗炎因子的平衡。枸杞多糖预处理可以通过抑制TXNIP的表达减少乙醇孵化后大鼠正常肝细胞系BRL-3A的细胞焦亡、氧化应激和NLRP3炎症介导的炎症反应,另一项研究表明,枸杞多糖也可以通过抑制NLRP3/6炎症途径和NF-κB的活化显著改善非酒精性脂肪性肝炎所致的肝损伤,使血清中谷丙转氨酶(glutamate-pyruvate transaminase,ALT)和谷草转氨酶(aspartate transaminase,AST)水平升高,改善肝脏氧化应激、纤维化、炎症和焦亡。灵芝多糖与枸杞多糖在肝功能的改善与治疗中具有相似的作用。在小鼠高氧诱导的急性肺损伤治疗中,枸杞多糖可以通过SIRT1依赖性抑制NLRP3炎症小体的激活,改善急性肺损伤,降低小鼠的死亡率。樟脑多糖可以使帕金森病(Parkinson’s disease,PD)小鼠纹状体多巴胺水平升高,NLRP3炎症细胞纹状体表达降低,明显改善小鼠的神经行为学。金银花多糖同样可以抑制NLRP3炎症小体的活化,降低IL-1β、Caspase-1的表达,对抑郁小鼠起到保护作用。金银花多糖与樟脑多糖在改善小鼠神经行为学中具有相似的作用。黄芪多糖能够通过抑制NLRP3炎症小体的激活,从而降低Caspase-1、IL-18和IL-1β的表达,对DSS诱导的结肠炎起治疗作用。对于多糖对NLRP3炎症小体的调节作用大多停留在调节炎症方面,均未深入研究对细胞焦亡的作用,因此对于多糖通过调节NLRP3炎症小体进而影响细胞焦亡仍需要进一步研究。
3.2 多糖对细胞焦亡其他途径的作用
在细胞焦亡其他途径中,Caspase-3起着至关重要的作用,Caspase-3作为细胞凋亡信号途径中的关键蛋白其活化机制已经被阐明。其中线粒体损伤诱导ROS过量产生是Caspase-3活化的途径之一,也是关键一环。因此下文通过探讨多糖对ROS的作用间接探讨多糖对焦亡其他途径的作用。
细胞内ROS水平与多种疾病的症状和表现密切相关。值得注意的是,线粒体受损会促进线粒体ROS的产生,从而导致总细胞内ROS的水平升高。而肿瘤细胞的代谢方式变化也会使得肿瘤细胞中ROS水平维持在高水平状态,因此通过调节ROS水平达到抑制肿瘤的效果成为可能。多糖可以提高肿瘤细胞中ROS水平,造成线粒体损伤,进一步诱导Caspase-3的活化,激活线粒体凋亡途径,进而起到抑制肿瘤生长作用。岩藻多糖通过促进ROS的产生,增加线粒体膜的通透性,降低Caco-2细胞的增殖能力,促进Caco-2细胞的凋亡。树舌子实体多糖通过MAPK/细胞外调节蛋白激酶(extracellular signal-regulated protein kinases,ERk)信号通路激活线粒体信号途径,增加ROS的含量,激活MCF-7细胞凋亡途径,产生抗肿瘤作用。另一项实验同样采用树舌子实体多糖作用MCF-7细胞并得出同样的结论,进一步验证了树舌子实体多糖可以通过增加MCF-7细胞中的ROS水平,激活细胞自噬,表现出较好的抗肿瘤活性。
4 结 语
细胞焦亡是机体与生俱来的自我保护和应对有害物质入侵的应答反应。适度的焦亡有助于巨噬细胞清除病原体、维持机体稳态。但是,持续的细胞焦亡会持续产生促炎细胞因子,使得组织长时间处于炎症状态,诱发多种疾病。已经发现细胞焦亡参与多种疾病的发生与发展,因此,通过调节细胞焦亡治疗各类疾病已经成为研究热点。在肿瘤治疗过程中,通过诱导细胞焦亡的发生可以明显抑制肿瘤细胞的生长。已有研究发现,小分子物质可以通过激活细胞焦亡经典途径以及焦亡其他途径起到抑制肿瘤的目的。虽然天然产物多糖在细胞焦亡抗肿瘤方面仍鲜报道,但在以往多糖抗肿瘤的研究中发现存在Caspase-3介导的细胞凋亡,并且Caspase-3也可以激活焦亡蛋白GSDME,诱导细胞焦亡。因此可以推测出,多糖在焦亡抗肿瘤方面有一定作用。细胞焦亡因其促炎的特性而成为引发各类疾病的主要诱因。因此通过抑制或减弱细胞焦亡也是控制疾病的潜在方法。炎症小体作为细胞焦亡过程中炎症因子释放的主要承担者,也是细胞焦亡过程的关键核心。多糖可以通过调节NLRP3炎症小体的活化起到调节炎症的作用,但是多糖是否可以进一步影响细胞焦亡仍需要进一步探究。
从相关报道中发现,调节细胞焦亡的靶点主要集中在NF-κB/NLRP3/Caspase-1/GSDMD、Caspase-4/5/11、ROS/Caspase-3/GSDME等信号通路以及其他Gasdermin家族蛋白。细胞焦亡各途径的关联仍不十分清楚,需要更多的研究加以阐明。而细胞焦亡与细胞凋亡以及细胞坏死之间存在着相互的串扰,三者在细胞死亡过程中的关联仍需要探讨。多糖能否与细胞表面某一特定受体结合,影响NF-κB/NLRP3/Caspase-1/GSDMD、Caspase-4/5/11、ROS/Caspase-3/GSDME上游的某一蛋白,从而激活焦亡相关通路有待进一步研究。因此,多糖对细胞焦亡的调控作用有待于更深入的研究。
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22 00:24:52
材料与冶金学报(2022年2期)2022-08-10 09:15:38
医学综述(2022年7期)2022-04-19 12:31:12
昆明医科大学学报(2020年12期)2021-01-26 00:44:42
作文成功之路·小学版(2020年6期)2020-07-27 01:48:28
世界科学技术-中医药现代化(2020年2期)2020-07-25 02:06:26
中国组织化学与细胞化学杂志(2017年1期)2017-06-15 20:27:43
西南军医(2015年3期)2015-04-23 07:28:32
国际心血管病杂志(2015年5期)2015-02-27 12:11:34
现代检验医学杂志(2014年6期)2014-03-03 02:14:24
