
密码子优化人PD-L1基因在巴斯德毕赤酵母中的表达与鉴定
2022-06-24 04:36战映璇王环汤潜玄超于建立张霞王清
精准医学杂志 2022年4期
战映璇 王环 汤潜 玄超 于建立 张霞 王清
(1 青岛大学医学部,山东 青岛 266071; 2 青岛大学附属医院检验科;3 中国科学院青岛生物能源与过程研究所; 4 深圳市国创纳米抗体技术有限公司)
程序性死亡配体-1(PD-L1)广泛存在于多种细胞细胞表面[1],与程序性死亡受体-1(PD-1)相互作用可以抑制T细胞活性与增殖,诱导免疫耐受[2]。这一过程在感染、肿瘤、自身免疫性疾病的发生发展中起重要的调控作用[3-6],PD-L1也是预测肿瘤免疫治疗的潜在生物标志物[7-11]。目前,实验室使用的商品化PD-L1蛋白主要来源于哺乳动物细胞系,也有使用大肠杆菌表达PD-L1的报道。然而,哺乳动物细胞培养条件苛刻,使商品化的PD-L1价格高昂;大肠杆菌作为原核细胞,无法对蛋白质进行翻译后加工修饰,并且表达的PD-L1以无活性的包涵体形式存在于细胞内,纯化难度高[12]。巴斯德毕赤酵母(Pichiapastoris,P.pastoris)为真核生物,其含有的细胞器可以帮助蛋白质折叠和糖基化修饰。另外,P.pastoris营养要求低,能够实现高密度发酵,蛋白表达后可分泌至培养基上清液中,提取纯化方便[13]。本研究拟通过基因工程手段,构建并筛选能稳定表达人PD-L1蛋白的重组P.pastoris,为开展PD-L1的相关研究奠定基础。
1 材料和方法
1.1 材料
P.pastorisGS115(武汉普因特生物工程有限公司);含有pPIC9K-PDL1质粒的XL10-Gold大肠杆菌(上海生工生物工程股份有限公司构建质粒并转化);质粒大提试剂盒(北京天根生化科技有限公司);卡那霉素(北京索莱宝科技有限公司);G418(美国Inalco公司);SacⅠ-HF(美国New England Biolabs有限公司);抗PD-L1单克隆抗体28-8(英国Abcam公司);电穿孔仪(美国Bio-Rad公司),胰蛋白胨、酵母提取物(美国Thermo Fisher Scientific公司)。本研究涉及的BMGY、BMMY、LB、YPD培养基,均按照巴斯德毕赤酵母表达手册(美国Invitrogen公司)配制。
1.2 方法
1.2.1目的基因的优化及重组质粒的构建 登录NCBI官网(https://www.ncbi.nlm.nih.gov/),查询到PD-L1基因的cDNA序列(PD-L1 transcript variation1,NM_014143.3),按照P.pastoris的密码子使用偏好优化基因序列,将序列中不常用的稀有密码子替换为常用的最优密码子,以提高密码子适应指数(CAI),剔除可能会限制mRNA的翻译、加速mRNA降解的不良序列。将优化后的基因序列人工合成,然后插入pPIC9K质粒,构建重组质粒pPIC9K-PDL1。将pPIC9K-PDL1转化至大肠杆菌XL10-Gold中,参照质粒大提试剂盒说明书从大肠杆菌中提取质粒。使用PCR鉴定提取到的质粒是否含有PD-L1基因,PCR引物为alpha-Factor:5′-TACTATTGCCAGCATTGCTGC-3′,AOX1:5′-GGCAAATGGCATTCTGACATCCT-3′。PCR反应条件为:95 ℃预变性2 min;95 ℃变性30 s,55 ℃退火30 s,72 ℃延伸1 min,共35个循环;最后以72 ℃延伸5 min。质粒鉴定无误后使用SacⅠ-HF将质粒线性化,线性化的质粒使用乙醇沉淀后回收,用于后续P.pastoris的转化。
1.2.2重组P.pastoris菌株的构建与鉴定 将线性化的质粒与感受态P.pastoris菌液混合,设置电转化参数:电压1.5 kV,电容25 μF,电阻200 Ω,电击时间4~10 ms,进行电击。电击后的菌液接种至无组氨酸的MD平板上,30 ℃培养3 d。为了高效筛选高拷贝的重组P.pastoris菌株,将MD平板上的菌落刮下,重悬于无菌水中,稀释后接种于G418浓度分别为0.5、1.0、2.0、3.0 g/L的YPD平板上,30 ℃培养72~96 h,在G418浓度大于0.5 g/L的平板上挑选菌株进行PCR鉴定,以质粒pPIC9K-PDL1的PCR产物作为对照。PCR引物和反应程序同步骤1.2.1。PCR结果阳性的菌株送至擎科生物科技有限公司测序,测序结果与优化后的PD-L1基因一致即为重组P.pastoris菌株。
1.2.3重组P.pastoris菌株的诱导表达 将重组P.pastoris菌株接种于5 mL YPD培养基中,30 ℃下摇床200 r/min培养24 h。然后再将菌株转接至30 mL BMGY培养基中,30 ℃下摇床200 r/min培养至培养基A600 nm达到2~6。离心收集菌体,将菌体重悬置于50 mL含有体积分数为0.005甲醇的BMMY培养基,使菌液A600 nm=1,开始诱导表达。分别于第24、48、72、96、120小时各留取1 mL诱导表达的培养基上清液备用,并补充甲醇至体积分数为0.005。
1.2.4表达产物的免疫印迹分析 使用三氯乙酸-丙酮法提取上清液中的蛋白,进行蛋白电泳。将电泳后的蛋白质转移至聚偏二氟乙烯(PVDF)膜上,先用含质量分数0.05的牛血清白蛋白封闭,使用抗PD-L1单克隆抗体28-8作为一抗,4 ℃孵育过夜。然后使用辣根过氧化物酶标记的山羊抗兔IgG作为二抗,室温孵育1 h,使用化学发光底物对表达产物进行显影。使用Image J软件分析免疫印迹检测结果中PD-L1蛋白条带的灰度值,对蛋白含量进行相对定量,寻找上清液中PD-L1含量最高的时间点作为最佳收获时间。使用Quantity one软件,建立蛋白marker中各条带的相对分子质量与条带迁移距离的点对点半对数曲线,得到蛋白的相对分子质量。利用肽和蛋白质分子量计算器(https://www.aatbio.com/tools/calculate-peptide-and-protein-mo-lecular-weight-mw),根据蛋白质氨基酸序列计算未经修饰的PD-L1蛋白的相对分子质量。
2 结 果
2.1 目的基因的优化及重组质粒的构建
PD-L1基因优化后,基因序列的CAI从0.71升高到0.96,优化过程中剔除了53~58位核苷酸的“ATTTA”和528~534位核苷酸的“GGTAAG”序列。优化前后的基因序列变化及密码子使用频率改变情况见图1,图中密码子的颜色表示该密码子在P.pastoris中的相对使用频率。使用频率低的稀有密码子用蓝色表示,使用频率高的最优密码子用红色表示。与优化前的基因序列相比,优化后的基因序列中稀有密码子(蓝色)大部分被最优密码子(红色)所取代。
重组质粒的PCR鉴定结果如图2A所示,泳道内仅出现单一条带,大小约1 300 bp,与目的基因预计扩增片段的分子量相符,说明目的基因已整合到质粒中,重组质粒构建成功。
2.2 重组P.pastoris的构建与鉴定
挑选了7株菌株,其PCR鉴定结果如图2B所示,各菌株的PCR产物在泳道1~2、4~8皆在约1 300 bp处产生单一条带,条带大小与泳道9阳性对照(质粒pPIC9K-PDL1的PCR产物)相同,初步证明该菌株为重组P.pastoris。菌株的基因测序结果如图3所示,箭头标注范围与优化后的PD-L1基因序列一致,确定为重组P.pastoris。

图中密码子的颜色表示该密码子在P.pastoris中的相对使用频率,颜色越偏向蓝色代表该密码子在P.pastoris中的使用频率越低,越偏向红色则代表使用频率越高
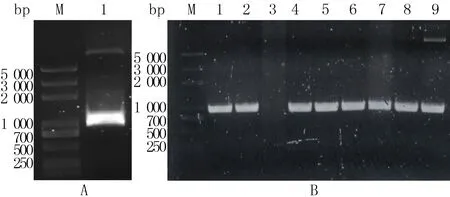
A:重组质粒PCR鉴定结果,M为DL2000 marker,1为质粒pPIC9K-PDL1的PCR产物;B:重组P.pastoris的PCR鉴定结果,M为DL2000 plus marker,1~2与4~8为7株重组P.pastoris的PCR产物,3为阴性对照,9为阳性对照(质粒pPIC9K-PDL1的PCR产物)
2.3 表达产物的免疫印迹分析
选取表达量相对较高的1株重组P.pastoris菌株,对其培养基上清液进行免疫印迹检测,结果如图4,重组PD-L1可以与抗PD-L1抗体特异性结合。其中,诱导第24小时的培养基上清液中未检测到PD-L1蛋白。第72小时培养基上清液中蛋白含量达到高峰,在第96小时蛋白含量下降。第72、96、120小时的表达量分别是第48小时的2.14、0.39、0.23倍。经Quantity one软件计算,重组PD-L1蛋白的相对分子质量约为44 700。经肽和蛋白质分子量计算器计算,PD-L1蛋白未经修饰状态下的相对分子质量为33 275。
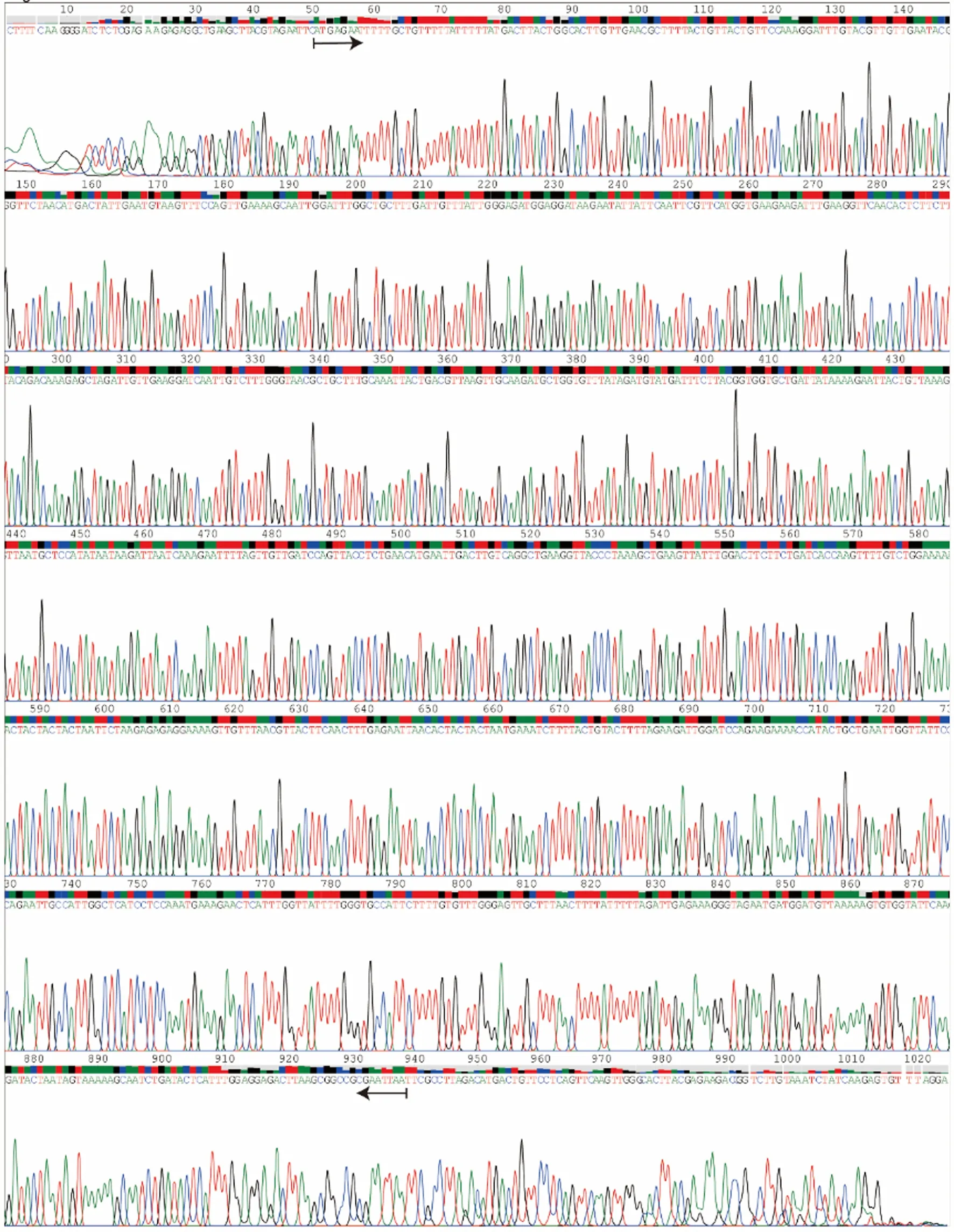
箭头标注范围内的序列与优化后的PD-L1基因序列一致,说明菌株为携带PD-L1基因的重组P.pastoris
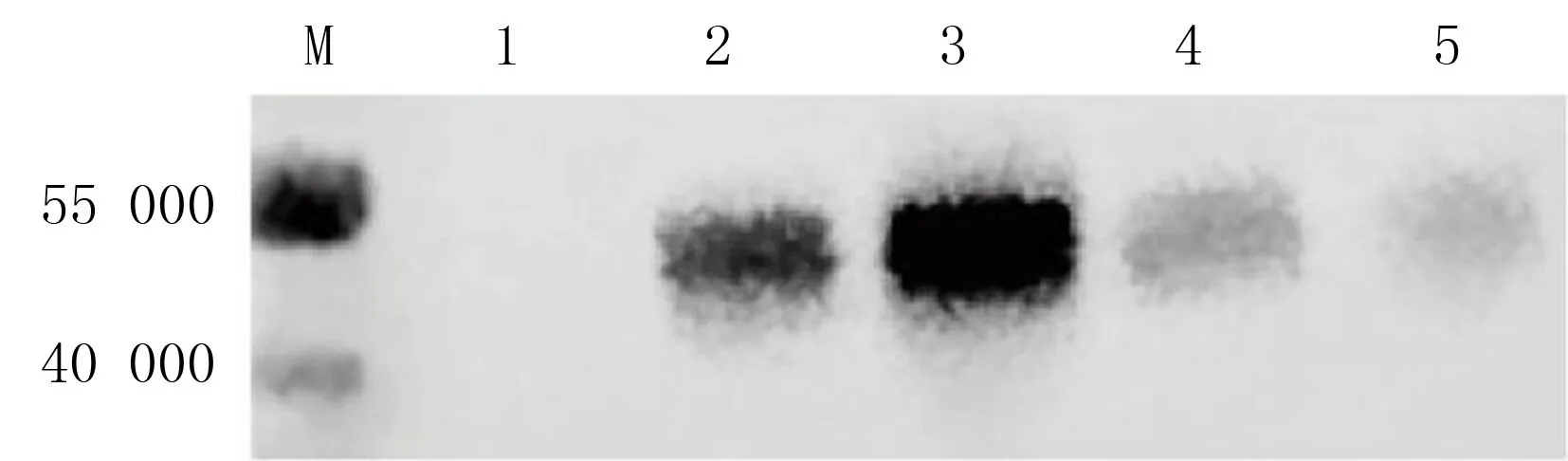
M为marker,1~5为诱导表达第24、48、72、96、120小时的培养液上清液中PD-L1蛋白的表达
3 讨 论
PD-L1是由290个氨基酸组成的Ⅰ型跨膜蛋白,其结构包括胞外结构域、跨膜结构域、胞内结构域[8]。PD-1、抗PD-L1单抗结合于PD-L1胞外结构域中的IgV结构域[8]。由于以往的研究大多关注PD-L1与PD-1、抗PD-L1胞外结构域抗体的结合,所以目前商品化的PD-L1主要为PD-L1蛋白的第19~238位氨基酸,即去掉N末端肽的胞外结构域,有关PD-L1蛋白表达的研究也多集中于此结构域。而最近的研究显示,PD-L1的胞内结构域介导的信号传导可降低干扰素对肿瘤细胞的毒性,而此过程不依赖于PD-L1胞外结构域与PD-1间的结合[14];在免疫组织化学(IHC)检测中,检测PD-L1胞内结构域或胞外结构域都可以反映病人预后,两者的结果具有一致性[15]。故本研究表达的PD-L1蛋白具有胞内结构域和胞外结构域,比起仅具有胞外结构域的PD-L1蛋白,其可以更广泛地应用于科研领域。
现有商品化PD-L1蛋白主要来源于人胚肾细胞HEK 293细胞系,作为哺乳动物细胞系,HEK 293表达的人源蛋白与天然蛋白更接近,但是由于培养条件苛刻,使用HEK 293细胞系表达人PD-L1蛋白需要承担高昂的生产成本。截至目前,已有凝血因子ⅩⅢa、干扰素α2b、人免疫球蛋白G的CH2结构域等多个人源蛋白在P.pastoris中成功表达并经验证具有生物活性[16-18],并且P.pastoris的营养要求低,可以利用甲醇诱导实现高密度发酵,相比哺乳动物细胞,可大大减少蛋白的生产成本。有学者曾探索使用大肠杆菌表达系统获得人PD-L1蛋白[12],由于大肠杆菌缺乏支持蛋白折叠的细胞器,其表达的PD-L1以包涵体形式存在于细胞内,需要通过多步的复性才能得到有活性的PD-L1。天然的PD-L1具有N35、N192、N200和N219四个N糖基化位点。这些糖基化位点位于IgC和IgV样结构域位置[8],对于PD-1和PD-L1的相互作用至关重要[19];IgC和IgV还是PD-L1 IHC抗体22C3和28-8的结合部位,去除N糖基化的PD-L1与抗PD-L1间的亲和性下降[20],所以,使用大肠杆菌来源的重组PD-L1免疫动物后,得到的抗体与人源PD-L1亲和性不高[19,21]。而P.pastoris作为真核生物,含有内质网和高尔基体,可以对蛋白进行折叠和翻译后的修饰。本研究表达的人PD-L1全长蛋白的相对分子质量约为44 700,与45 000的肿瘤细胞来源的PD-L1[19]相对分子质量相似。而通过氨基酸序列计算得出的人PD-L1蛋白的相对分子质量为33 275,因此认为两者的相对分子质量差异来源于蛋白质翻译后的加工修饰。关于PD-L1蛋白免疫原性的探讨,我们计划在后续的研究中使用PD-L1蛋白免疫羊驼,验证蛋白的免疫原性,进一步筛选抗PD-L1纳米抗体。
优化编码目的蛋白基因是提高外源蛋白表达量的策略之一[22-23]。CAI可以反映目的基因密码子与表达系统偏好的密码子相对使用频率的符合程度[24],本研究按照P.pastoris表达系统的偏好,在保证重组蛋白氨基酸序列不变的情况下,对目的基因进行优化,使CAI从0.71提高到0.96。本研究还在优化的过程当中剔除了可能导致mRNA降解的“ATTTA”序列[25],和可能导致mRNA降解和基因沉默的“GGTAAG”序列[26-27],以保证目的基因转录和目的蛋白翻译效率。
诱导表达不同时间点的目的蛋白的相对含量分析显示,重组P.pastoris菌株诱导表达的72 h内,上清液中蛋白含量呈上升趋势,第96小时则检测到蛋白含量下降。后续可在诱导表达的第72小时收集培养基上清液,以实现蛋白产量的最大化。诱导后期上清液中蛋白含量下降,可能是由于发酵过程中细胞密度过高,部分细胞裂解,胞内蛋白水解酶释放而导致PD-L1的降解。针对这一问题,在后续的研究中可向培养基中添加合适的蛋白酶抑制剂和蛋白酶水解底物,以减少蛋白的降解。另外,可将菌株接种于发酵罐,优化pH、温度等发酵条件[28-29],为菌株提供更稳定、更适宜的生长环境,以进一步提高蛋白的表达量。
猜你喜欢
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
成都医学院学报(2022年4期)2022-08-19
湖北农业科学(2022年11期)2022-07-18
福建农业学报(2021年6期)2021-08-18
江西农业学报(2021年4期)2021-04-20
实用肿瘤学杂志(2020年4期)2020-12-08
教学考试(高考生物)(2020年4期)2020-11-18
三农资讯半月报(2020年11期)2020-06-21
发明与创新·中学生(2019年6期)2019-06-26
安徽农业科学(2018年1期)2018-05-14
