
变分自编码器对甲基化缺失数据的填补
2022-06-23 06:24王新峰
计算机工程与应用 2022年12期
王新峰,黄 伟
1.吉首大学 软件学院,湖南 吉首 416000
2.中山大学 计算机学院,广州 510000
3.中南大学 计算机学院,长沙 410000
DNA甲基化(DNA methylation)是表观遗传基因组中重要组成部分,它能在不改变DNA序列的前提下改变遗传表现,也是研究最深入的表观遗传调控机制之一。DNA甲基化是指DNA序列上特定的碱基在DNA甲基转移酶(DNA methyltransferase,DNMT)的催化作用下,以S-腺苷甲硫氨酸作为甲基供体,通过共价键结合的方式获得一个甲基基团的化学修饰过程[1]。它在基因表达和调控中起着重要作用,并参与许多细胞过程,包括细胞分化、发育和肿瘤发生。最常见的DNA甲基化类型为5-甲基胞嘧啶(5mC)、6-甲基腺嘌呤(6mA)和4-甲基胞嘧啶(4mC),其中5mC是研究最广泛的类型[2]。很多研究表明DNA甲基化的变化与多种疾病的发病机制有关。例如,在许多癌症类型中,肿瘤细胞通常表现出与健康细胞不同的DNA甲基化模式,而肿瘤抑制基因很可能因启动子区域的高甲基化而失活[3]。CpG岛(CpG island)的高甲基化与肾上腺皮质癌(Adrenocortical carcinoma,ACC)癌症的低存活率有关[4]。近年来高通量基因测序技术虽取得了巨大进步,但DNA甲基化测序的序列数据因为各种技术局限性还常包含很大一部分缺失值导致无法直接使用[5]。因此有必要对这些序列数据中的缺失值进行估算填补。
缺失数据可以分为三种类型[6]:(1)完全随机缺失(missing completely at random,MCAR),缺失情况与任何变量无关;(2)随机缺失(missing at random,MAR),缺失的可能性不取决于缺失值,而是取决于观察值;(3)非随机缺失(missing not at random,MNAR),缺失情况只发生在特定的缺失数据上。填补方法需要对缺失数据的类型进行假设,没有研究明确指出DNA甲基化的数据缺失类型。根据已有文献的假设和实验经验,假设DNA甲基化的缺失类型为MCAR或MAR。已有一些基于统计和机器学习的方法被应用于随机缺失值的填补。如最近邻KNN(K-nearest neighbor)方法通过选择与样本最相似的K个样本,计算这些样本的平均值用于填补该样本的缺失部分。郝胜轩等[7]基于KNN使用所有非噪声最近邻对缺失数据进行填补取得很好效果。主成分分析(principal component analysis,PCA)[8]和奇异值分解(singular value decomposition,SVD)方法[9]基于相同原理都是通过计算数据矩阵的协方差矩阵,获得特征值的特征向量,通过特征向量可以生成新的矩阵用于缺失数据的填补。
深度学习是基于数据表示的机器学习方法,通过组合低级特征形成更加抽象的高级表示特征,以发现数据的分布式特征[10]。它强大的建模复杂非线性关系的能力在许多领域已显示出极大的优越性[11],如语音识别[12]、图像识别[13]和自然语言处理[14]等领域。近年来,深度学习的发展已被越来越多的应用于生物信息领域,如蛋白质结构预测[15-17]、药物设计[18-20]、基因组分析[21-23]和单细胞基因组分析[24-27]。自编码器(Auto-Encoders,AE)是深度网络中一种常见的模型,具备生成与训练数据相似数据样本的功能,可应用于数据增广和填补。已有研究者将转移学习技术与自编码器相结合来插补缺失的RNA序列数据[28]。变分自编码器[29](variational auto-encoder,VAE)是深度隐含空间生成模型的一种,通过学习隐含编码空间与数据生成空间的特征映射,进而在输出端重构生成输入数据。它是无监督特征学习的重要工具,在图像生成方向取得了极大的成功[30]。正是VAE这种强大的重构输入数据的能力,为DNA甲基化缺失数据的填补提供了新的机遇。
1 填补实验材料
1.1 数据集
基准实验数据集选自The Cancer Genome Atlas(TCGA)平台[31],包含信息有:DNA甲基化数据(JHU-USC Human Methylation 450类型)、临床信息和后续随访信息。数据集中共有33种不同的癌症样本9 756个,每个样本具有485 577个甲基化位点。DNA甲基化水平通过计算甲基化和未甲基化等位基因之间的强度比来确定,称为β值(范围从0到1)。数据样本中的部分位点在所有样本上都缺失,这样的缺失位点没有填补价值在填补之前先删除。
TCGA平台上不同癌症样本分布极为不均从几十到几百不等,考虑到深度学习需要的样本量和癌症的代表性,最终从平台中挑选了发病率很高的肺癌(lung adenocarcinoma,LUAD)作为填补实验样本,LUAD含有507个样本。
1.2 模拟缺失值
由于癌症样本数据中缺失位置对应的真实值无法获取,则不能对填补值的准确性进行直观评估。为了更好地评估不同填补方法的填补性能,需要在模拟缺失值上进行仿真实验。主要流程为:(1)从LUAD数据集中选择方差最大的前10 000个无缺失位点形成仿真数据集D∈R507×10000(507为样本数量,10 000为每个样本的甲基化位点数量)。(2)在仿真数据集中随机引入不同的缺失比例(取20%、40%、60%、80%),并用相同大小的矩阵记录下缺失位置信息。(3)对引入缺失值的数据进行填补。图1为在D∈R5×5矩阵的每列随机引入20%缺失值的流程。原始矩阵大小为[5,5],每列引入1个(5×0.2)缺失值,将该位置数值置为0。同时,在标记矩阵中的相同位置置为1代表引入的缺失值位置。填补操作完成后,填补矩阵中X͂ij为填补后的值。通过标记矩阵中1的位置,在原始矩阵和填补后矩阵中找到填补前后的值就可进行准确性评估。
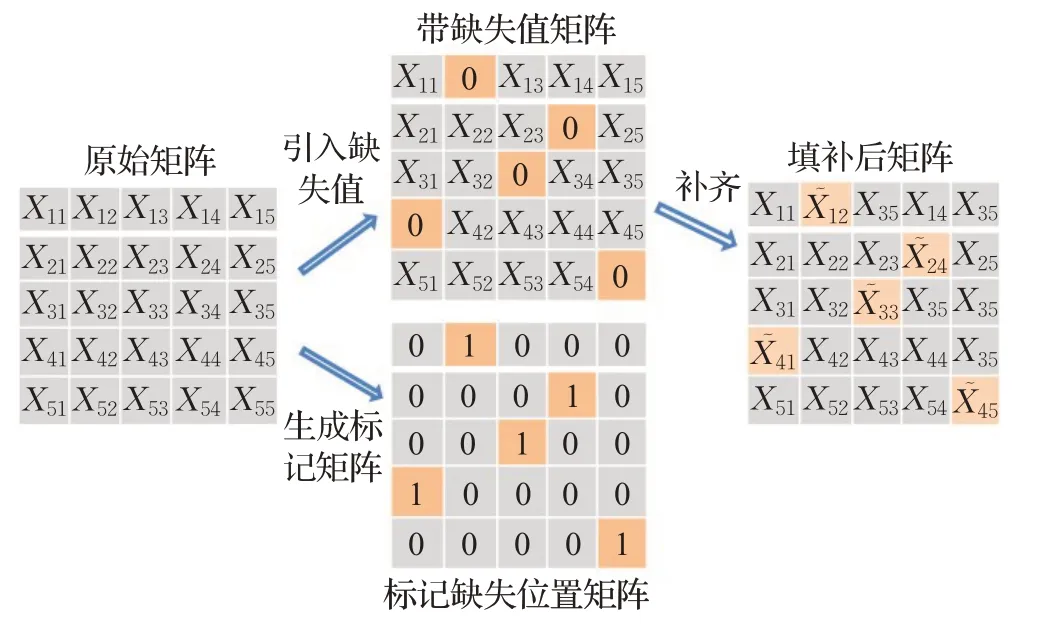
图1 模拟缺失值引入流程Fig.1 Simulate process of introducing missing values
2 填补实验
填补实验主要分为三个部分:(1)基于变分自编码器原理建立用于填补DNA甲基化缺失值的模型VAEMethImp。(2)将VAE-MethImp模型与现有的方法进行对比,通过均方根误差(root mean squared error,RMSE)和拟合度(R-squared,R2)对填补值的精度进行评估。(3)通过生存分析实验来验证填补值的真实有效性。
2.1 变分自编码器模型结构
自编码器模型主要有两个部分组成:(1)编码器(Encoder):学习输入数据的隐含特征的空间表示;(2)解码器(Decoder):从学习到的低维特征中重构出原始的输入数据。如图2所示,编码器h将原始输入X编码运算得到特征X',X'=h(X);解码器f将特征X'解码生成X͂,即:X͂=f(X')=f(h(X))。X和X͂越接近意味着提取的X'特征代表性越好。
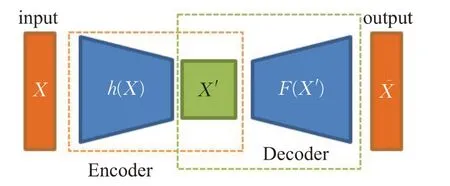
图2 自编码器结构示意图Fig.2 Structure diagram of auto-encoder
变分自编码器除了拥有与图2中自编码器结构相似的编码器和解码器外,还新增了隐含编码,可以看作是神经网络和贝叶斯网络的混合体。在VAE中,隐藏编码对应的结点可以看成是随机变量,其他结点视为普通神经元。编码器功能是一个变分推断网络,用来推荐均值和方差,而解码器可以看作是将隐变量映射到观测变量的生成网络。VAE-MethImp模型如图3所示,模型共分为3部分:
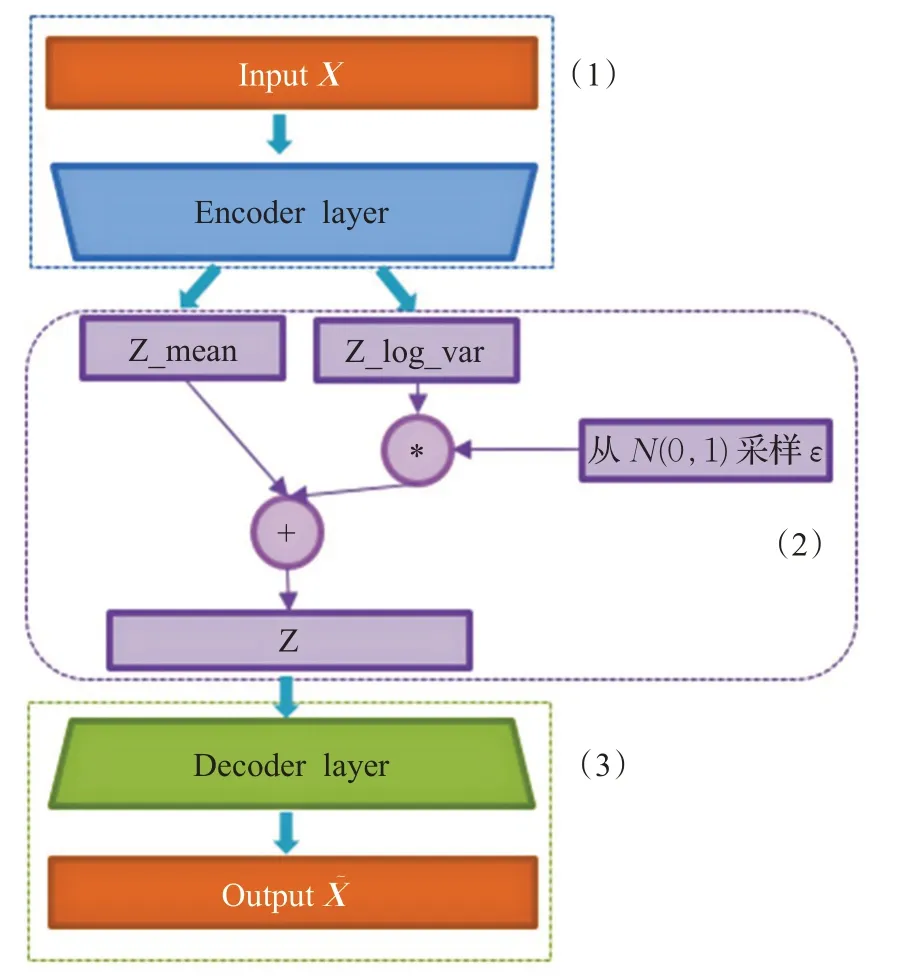
图3 VAE-MethImp模型结构图Fig.3 VAE-MethImp model structure diagram
(1)编码层:编码层从输入X推断出分布的均值和方差,功能与自编码器中的Encoder功能一致,Z_mean,Z_log_var=h(X)。
(2)隐含变量Z:Z是通过编码层输出的均值和方差计算出的输入数据的专属正态分布。
(3)解码层:Z通过解码层生成重构后的数据X͂。
编码层用来推断Z的均值和方差,再从正态分布中取采样ε,通过计算得到Z,如公式(1)所示:

其中zi∈Z,mi∈Z_mean,σi∈Z_log_var。
VAE-MethImp模型训练的目标有两个:编码层需要将推断出的正态分布与标准正态分布的KL散度KL(N(m,σ2)||N(0,I))作为额外的Loss,最小化损失函数如公式(2)所示:

其中xi∈X,x͂i∈X͂。
超参数选择是训练深度神经网络模型中重要的环节,选择一组最优超参数可以提高学习的性能和效果。通过网格搜索对最常见的7种超参数进行尝试和优化,最终选择的超参数集如表1所示。
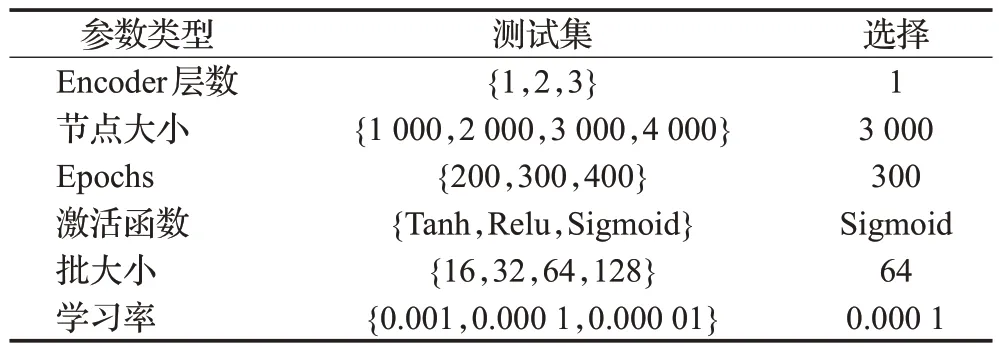
表1 测试的超参数集与选择Table 1 Tested and selected hyperparameters
2.2 对比方法的实施
对现有的填补方法进行了分析与比较,确定了4种常见的填补方法,分别是Mean、KNN、imputePCA和SVD。随机森林和多重填补法(MI)由于处理[507,10 000]大小的矩阵所耗费的时间(超48 h未结束)无法接受被舍弃。在实现时imputePCA直接调用missMDA包实现,SVD实现由fancyimpute包提供。所有方法均使用默认参数实现,同时进行了一些关键参数的优化。具体说来,在KNN方法中邻居数K取值为30(从10、20、30和50中挑选),SVD的最大特征值设置为10(从5、10、20和30中挑选)。
2.3 生存分析
RMSE和R2只能用来评估填补值的平均准确性,无法衡量填补值的实际有效性。填补值的实际有效性可以用岭回归正则化的COX比例风险模型(cox proportional-hazards model)来预测。该模型使用R软件包中适用于高维数据拟合的glmnet包构建[32]。通过一致性指数(C-Index)和p值(p-value)两个指标对预测结果进行评估。C-Index是由Harrell教授提出用于计算生存分析中的COX模型预测值与真实之间的区分度[33],C-Index值为0.5即为随机猜测,数值越高则区分度越好。p值衡量将患者分为高风险组和低风险组的概率,值越低区分度越好。
3 结果与讨论
统计了VAE-MethImp和现有的4种方法(Mean、KNN、imputePCA、SVD)在LUAD仿真缺失数据集上的填补实验结果。通过RMSE、R2和C-Index三个指标对各方法填补后的数值精度和有效性进行了评估。
3.1 填补精度评估
图4中显示了VAE-MethImp方法与其他方法的填补结果对比,结果显示VAE-MethImp方法优于其他方法。具体来看,Mean和KNN拥有较高误差,KNN的RMSE随着缺失率的增大而迅速增加。当缺失率超过40%时,KNN的结果差于Mean,因为在高缺失率下,KNN通过仅存的部分信息获取的邻居之间相似度差。imputePCA与SVD原理相同,但imputePCA基于更少的特征向量导致RMSE比SVD高。VAE-MethImp方法对比SVD将误差平均缩小了4.8%,缺失率越低VAEMethImp的优势越大,在80%缺失率时两者接近原因是缺失数据太多可提取的特征变少。R2指标的趋势与RMSE相同,VAE-MethImp填补后的数值相关性最好。
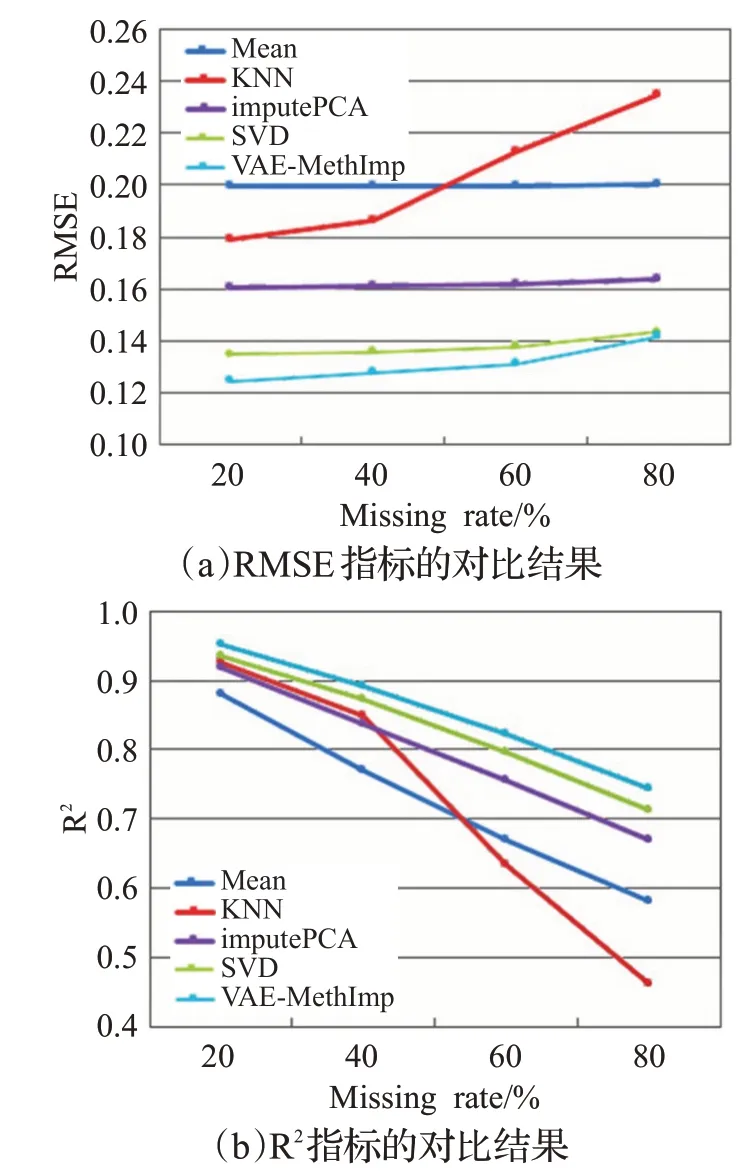
图4 5种填补方法的RMSE和R2结果对比Fig.4 Comparisons of five methods by RMSE and R2
除了填补性能更好外,VAE-MethImp方法在模型训练完后,对数据的填补花费时间也是属于最优行列。在对比新样本的填补时间时未将模型训练时间放入考虑是因为训练模型时间为一次性花费时间。在Titan 1080 GPU上测试对样本的填补时间与Mean方法接近,而KNN、imputePCA和SVD方法花费的时间取决于样本的大小,测试实验结果显示这些方法花费的时间是VAE-MethImp的10倍以上。
3.2 生存分析结果
为了验证填补数值的真实有效性,通过岭回归正则化Cox模型对不同填补方法的填补数值进行了生存分析。如图5所示,VAE-MethImp在4种缺失率下始终表现最佳。在缺失率为20%时,VAE-MethImp的C-Index为0.618,比Mean、KNN、imputePCA和SVD的结果分别提升10.5%、8%、4.7%和4%。很明显,所有填补方法的C-Index值都会随着缺失率的增加而减小,而填补方法之间的差异也会扩大。在缺失率为80%时,VAEMethImp比SVD方法的优势有所减少,与图4中的精度对比结果趋势相同。
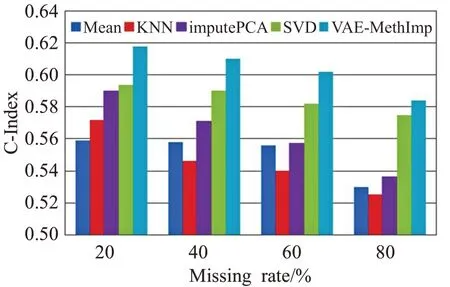
图5 5种方法的C-Index结果对比Fig.5 Comparisons of five methods by C-Index
3.3 LUAD和BRCA真实缺失数据实验
在模拟缺失数据集上进行的仿真实验表明,VAEMethImp模型在RMSE、R2和C-Index指标上都优于其他方法。为了进一步证明模型的鲁棒性,需要在真实缺失数据集上进行验证。除了已有的LUAD癌症,还增加了乳腺癌(Breast adenocarcinoma,BRCA)数据集进行真实填补实验,实验中包含的数据集如表2所示。首先,从LUAD和BRCA数据集中挑选缺失率最高的前10 000个位点,组成真实缺失数据集。然后,将5种方法分别对真实数据集进行填补,由于真实缺失位点对应的真实值无法获得,故RMSE和R2指标不能使用。最后,通过C-Index和生存曲线来查看不同方法的填补性能。图6所示为5种填补方法填补后的数据集和原始未填补数据集(No impute)的C-Index。图中显示,未填补数据集的C-Index接近0.5,表明原始数据集由于缺失数据太多变成无效数据。而经过不同方法填补后的C-Index有了提升,证明填补后的数据是有价值的,其中VAE-MethImp比SVD有了平均2%的提升,比未补齐的数据提升了12%。
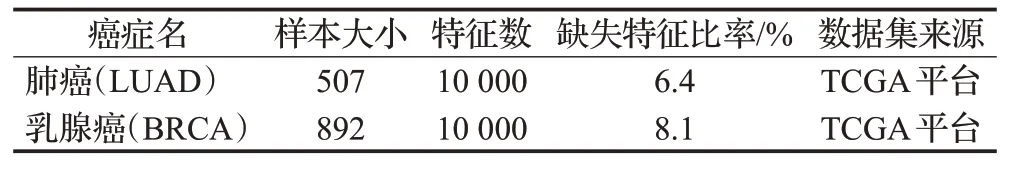
表2 实验数据集信息Table 2 Experimental dataset information
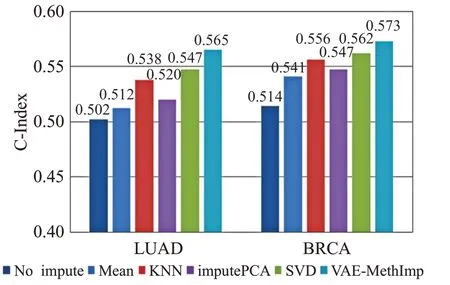
图6 不同填补方法在LUAD和BRCA癌症的生存分析结果Fig.6 C-Index value of survival prediction of original data(no imputation)and imputed methylation values on LUAD and BRCA
图7展示了经过VAE-MethImp填补后的数据与未填补数据的生存曲线对比,填补之后的数据集对高低风险组的区分比未填补的高低风险组的区分曲线间隔大,区分度更明显。两个癌症上的真实填补实验可以证明VAE-MethImp在填补性能上比其他方法更加优秀,填补之后的数据包含更多有价值信息。
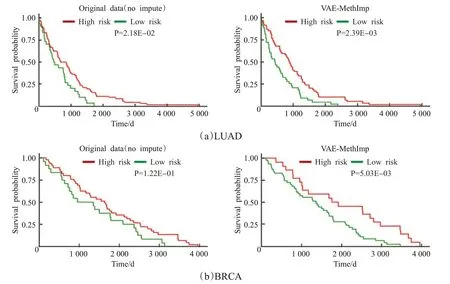
图7 LUAD和BRCA的填补前后生存曲线对比图Fig.7 Kaplan-Meier plots of predicted high-and low-risk patients by original and predicted values by VAE-MethImp for LUAD and BRCA
4 总结
在本项研究中,开发了一种新的基于变分自编码器的用于DNA甲基化缺失数据的填补方法,即VAEMethImp。利用VAE极强的重构生成输入数据的能力,有效地解决了DNA甲基化缺失的问题。通过在模拟的缺失数据上的仿真实验,证明VAE-MethImp在RMSE和R2方面均优于现有方法。通过对填补数据的生存分析进一步证实了填补数值的有效性。在LUAD和BRCA癌症的真实缺失数据集上的填补实验也表明,VAEMethImp方法比其他方法填补的数据,拥有更高的CIndex值和区分度更明晰的生存曲线。
目前,本研究仅限于癌症的DNA甲基化缺失数据的填补上,且只利用了单个癌症样本之间的相关性。后续可以考虑利用泛癌样本间DNA甲基化的相关性来提高单个癌症的数据填补精度。随着测序技术的发展,该填补方法可以进一步应用于其他生物组学类型、不同疾病类型及其他任务上,例如年龄预测和细胞分类。
猜你喜欢
网络安全与数据管理(2022年1期)2022-08-29
昆明医科大学学报(2022年1期)2022-02-28
上海金属(2021年6期)2021-12-02
昆明医科大学学报(2021年3期)2021-07-22
烟草科技(2021年6期)2021-06-24
科学技术创新(2021年5期)2021-03-17
中学生物学(2020年10期)2020-12-25
——编码器
演艺科技(2020年7期)2020-08-13
电脑知识与技术(2018年19期)2018-11-01
探测与控制学报(2015年4期)2015-12-15
