
国槐SSR-PCR反应体系的建立与优化
2022-06-13 09:42张明静庞彩红李际红李双云付茵茵哈新英夏阳
山东农业科学 2022年5期
张明静,庞彩红,李际红,李双云,付茵茵,哈新英,夏阳
(1.山东省林业科学研究院/山东省林木遗传改良重点实验室/黄河三角洲林木遗传育种国家林草局重点实验室,山东 济南 250014;2.山东师范大学生命科学学院,山东 济南 250014;3.山东农业大学林学院,山东 泰安 271018;4.济南市城乡建设发展中心,山东 济南 250000)
国槐(Sophora japonica L.)为豆科(Leguminosae sp.)蝶形花亚科(Papilionoideae)槐属(Sophora L.)落叶乔木,是我国北方重要的乡土树种和城市绿化树种,具有较高的观赏价值[1];其速生性较强,材质坚硬,花、枝、果实等均可入药[2],也是良好的材用和药用经济树种。目前,国内关于国槐的研究还主要集中在繁殖技术[3]、遗传转化[4]以及种子表型性状的变异[5]等方面,针对其分子方面的研究开展较少,严重影响国槐种质资源的创新和利用。
目前应用于植物遗传育种研究的分子标记技术主要有AFLP、RAPD、RFLP、SSR、ISSR等。SSR(simple sequence repeat)即简单重复序列,又称微卫星。通常采用聚丙烯酰胺凝胶电泳或高浓度的琼脂糖凝胶电泳对SSR-PCR产物进行检测,其结果可以显示出不同品种间重复次数带来的差异性[6]。SSR标记由于多态性高、数量丰富、在遗传上呈共显性、实验方法简单、结果相对稳定可靠等优点,被广泛应用于DNA指纹图谱构建[7,8]、分子辅助选择[9]和遗传多样性分析[10]等研究中,是目前应用比较广泛的一种分子标记技术。
本研究旨在利用单因素试验结合正交试验设计,建立一套适用于国槐SSR标记的稳定、可靠的PCR反应体系,以期为利用SSR分子标记技术对国槐进行遗传多样性分析、指纹图谱构建、种质资源鉴定等方面的研究奠定基础。
1 材料与方法
1.1 试验材料与试剂
选用国槐种质WF56、TA11、BZ26、DY17、ZB9、LY86、42、201、396,均为嫁接保存于山东省林业科学研究院饮马泉苗圃及章丘实验基地的国槐古树,具体源生地见表1。2016年春天,从国槐嫁接苗上采集嫩叶,清洗干净,放入2 mL离心管中,编号,液氮迅速冷冻后放入-50℃冰箱备用。
植物基因组DNA提取试剂盒购自北京天根公司;DNA Marker、ExTaq酶、10×Taq Buffer、MgCl2、dNTPs、6×Loading Buffer等均购自宝生物工程(大连)有限公司(TaKaRa);冰乙酸、硝酸银、无水乙醇购自天津市凯通化学试剂有限公司。

表1 国槐种质材料的源生地
利用高通量测序平台Illumina HiSeq 2000进行国槐转录组测序,测得7 Gb数据,进行数据denovo组装,共获得68 846个uigene,使用SSR软件MIcroSAtellite(MISA)寻找所有的SSR位点,对所有SSR重复单元在Unigene上前后序列的长度进行筛选,得到8 718条包含SSR位点的序列,共含有10 236个SSR位点,设计完成6 611条引物。从中选出226对引物由铂尚生物技术(上海)有限公司合成,每对引物5 OD,分装两管,-20℃冰箱保存备用。从226对引物中随机挑选5对引物(表2)用于本试验。

表2 选用的SSR-PCR引物及其序列
1.2 试验方法
1.2.1 国槐基因组DNA的提取与检测 采用植物基因组DNA提取试剂盒进行DNA提取。利用0.8%琼脂糖凝胶电泳检测提取的DNA质量,使用分光光度计测定提取的DNA浓度,根据分光光度计测定的DNA原液浓度将样品稀释至所需浓度,置于-20℃冰箱中保存备用。
1.2.2 PCR扩增程序及产物检测 采用Touchdown PCR扩增,程序如下:94℃5 min;94℃15 s,66.5~57.0℃15 s(每循环降0.5℃),72℃30 s,19个循环;94℃15 s,57℃15 s,72℃30 s,15个循环;72℃10 min,4℃保存。扩增后的产物利用8%非变性聚丙烯酰胺凝胶进行电泳,银染显色,相机拍照保存。
1.2.3 PCR扩增反应体系优化 随机挑选WF56国槐DNA为模板,利用2114引物进行PCR扩增,初始反应体系(10μL):5 U/μL ExTaq酶0.1μL,25 mmol/L Mg2+1.0μL,2.5 mmol/L dNTPs 0.4μL,10μmol/L引物0.2μL(上下游引物合计),70 ng/μL模板DNA 1.0μL,10×PCR Buffer 1.0μL,ddH2O补足到10μL。
采用单因素试验与正交试验相结合的方法对SSR-PCR反应体系中的Mg2+、引物、dNTPs、Ex-Taq酶、模板DNA用量五个因素进行优化。单因素试验中每个因素设置6个水平(表3),根据扩增条带清晰度确定各因素的适宜用量范围;再根据单因素试验结果,选用合适的正交试验设计表对5个因素的用量组合进行进一步优化,以确定适合国槐的10μL SSR-PCR最佳反应体系。
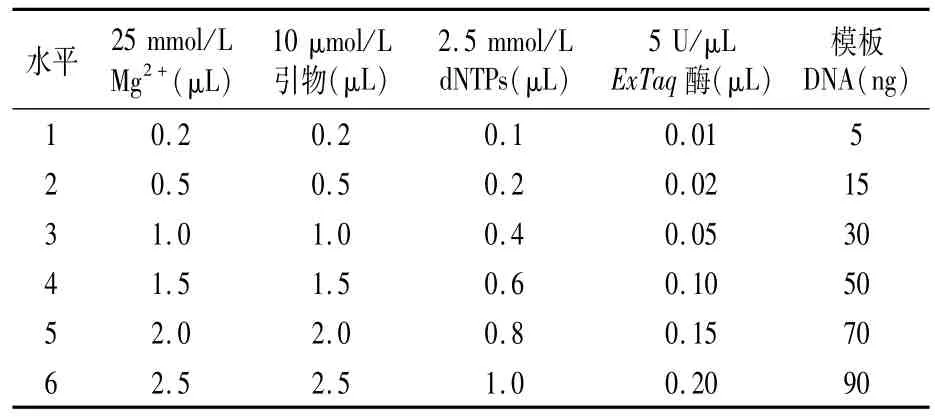
表3 国槐SSR-PCR反应体系单因素试验因素及水平
1.2.4 PCR优化体系的验证 随机挑选出8个国槐种质和4对SSR引物,利用筛选出的最佳反应体系进行PCR扩增,通过对扩增结果的分析对PCR反应体系进行有效验证。如果扩增出的条带清晰、稳定,则表明试验确定的反应体系适宜国槐SSR-PCR反应。
2 结果与分析
2.1 国槐DNA提取质量检测
经紫外分光光度计检测,提取的国槐基因组DNA的OD260/OD280值在1.7~2.0之间;经0.8%琼脂糖凝胶电泳检测(图1),条带清晰完整且无明显的拖尾。说明提取的DNA样品浓度和纯度较高,足以满足后续试验的要求。

M为DL15000 DNAMarker;1~9分别代表国槐种质WF56、TA11、BZ26、DY17、ZB9、LY86、42、201、396的基因组DNA。
2.2 单因素试验结果与分析
2.2.1 Mg2+用量对国槐SSR-PCR反应的影响
在PCR反应体系中,Mg2+与dNTPs相互作用影响酶的活性,进而影响PCR扩增效果[11]。Mg2+浓度过高会降低PCR扩增的特异性,浓度过低则可能使PCR扩增产物量减少甚至扩增不出条带[12]。如图2所示,Mg2+用量为0.2μL时未扩增出条带,用量为0.5μL时扩增出的条带较弱;用量为1.0~2.5μL时均扩增出清晰条带,但随着Mg2+用量的增加,条带的背景也呈逐渐加深趋势。综合考虑,Mg2+用量1.0μL较为适宜,扩增出的条带清晰且无明显背景。基于此,选择0.5~2.0μL的Mg2+用量用于后续正交试验分析。
2.2.2 引物用量对国槐SSR-PCR反应的影响

M指DL500 DNA Marker;1~6分别指25 mmol/L Mg2+用量为0.2、0.5、1.0、1.5、2.0、2.5μL。
引物浓度是影响PCR扩增结果的重要因素,浓度较低时很难进行有效扩增,往往扩增不出目的条带,而浓度过高则很容易引起错配或扩增出很多非特异性条带[11]。由图3可以看出,当引物用量在0.2μL时,扩增条带较弱,当引物用量达0.5 μL时,条带明显清晰,随着引物浓度的不断增加,条带的清晰度不断增加,但也逐渐出现弥散现象。因而用量最少且又扩增出清晰条带的0.5μL为引物适用量,并选择引物0.2~1.5μL用量范围用于正交试验分析。
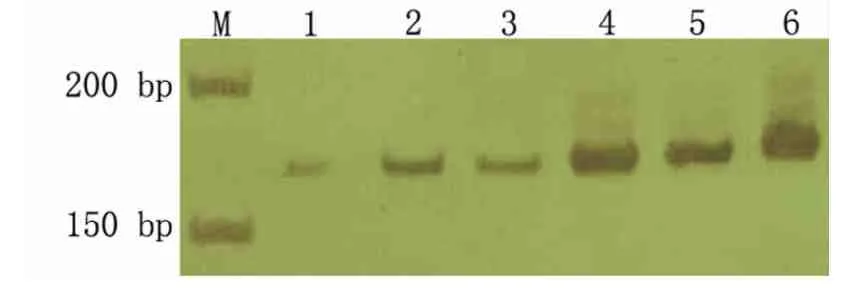
M指DL500 DNA Marker;1~6分别指10μmol/L引物用量为0.2、0.5、1.0、1.5、2.0、2.5μL。
2.2.3 dNTPs用量对国槐SSR-PCR反应的影响 dNTPs的浓度与PCR扩增效率有密切关系,浓度过低,PCR产物的产量减少,扩增出的条带不清晰;浓度过高,则会与Mg2+结合,降低游离Mg2+浓度,从而使酶的聚合效率降低,影响扩增结果[13]。由图4可知,当dNTPs用量在0.1μL时,扩增出的条带较弱,从0.2μL开始均扩增出清晰条带,且之后变化不大,因而0.2μL为较佳的dNTPs用量,选择0.2~0.8μL dNTPs用于正交试验分析。
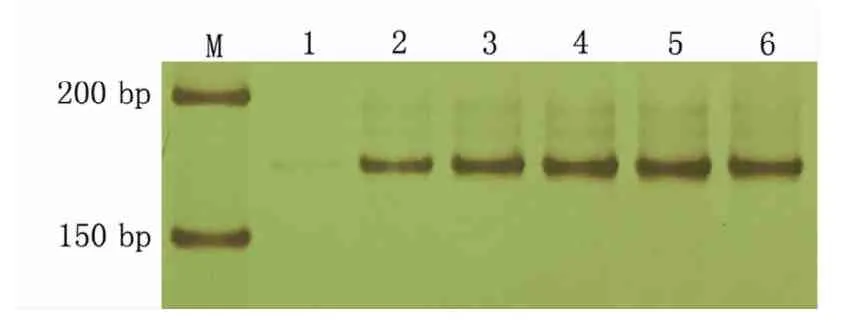
M指DL500 DNA Marker;1~6分别指2.5 mmol/L dNTPs用量为0.1、0.2、0.4、0.6、0.8、1.0μL。
2.2.4 ExTaq酶用量对国槐SSR-PCR反应的影响 ExTaq酶是PCR反应的关键,但浓度较高时会使非特异性产物增加,而浓度较低时则会使扩增量不足,扩增出的条带较弱[11]。由图5可以看出,ExTaq酶用量在0.01、0.02μL扩增出的条带微弱,用量继续增加,条带逐渐清晰,但同时背景也在逐渐加深。综合考虑,可以选择扩增条带清晰且无明显背景的0.05μL作为适宜的ExTaq酶用量。基于此,选择0.02~0.15μL ExTaq酶设计正交试验。
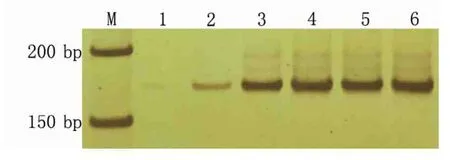
M指DL500 DNA Marker;1~6分别指5 U/μL ExTaq酶用量为0.01、0.02、0.05、0.10、0.15、0.20μL。
2.2.5 模板DNA用量对SSR-PCR反应体系的影响 如图6所示,模板DNA用量为5 ng和15 ng时,扩增出的条带极其微弱;当用量增加到30 ng时,能扩增出清晰的条带;用量为50、70 ng时,条带清晰度相对于30 ng时有所增加,但无较大区别;当用量增加到90 ng时,条带出现较弱的虚化现象。因而模板DNA用量为30 ng时较为适宜,在此基础上选择15~70 ng模板DNA用于后续正交试验设计。
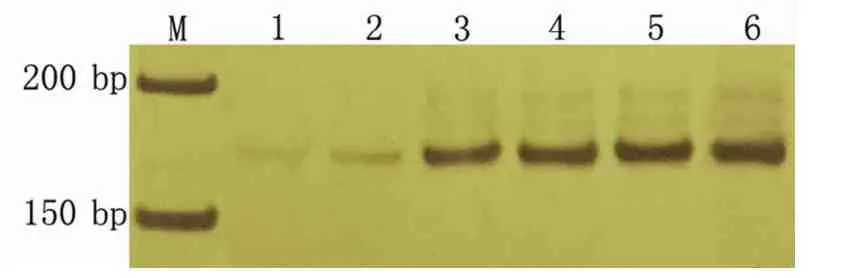
M指DL500 DNA Marker;1~6分别指模板DNA用量为5、15、30、50、70、90 ng。
2.3 正交试验结果
根据单因素试验选择的各因素用量范围,设计5因素4水平正交试验,选用L16(45)正交表设计因素组合(表4),然后利用引物2114对国槐WF56进行PCR扩增。
电泳结果(图7)显示,不同因素组合的PCR扩增结果存在较大差异,其中,组合1、2、12未扩增出条带,组合3、4扩增出的条带较弱,其余组合均扩增出清晰的条带。
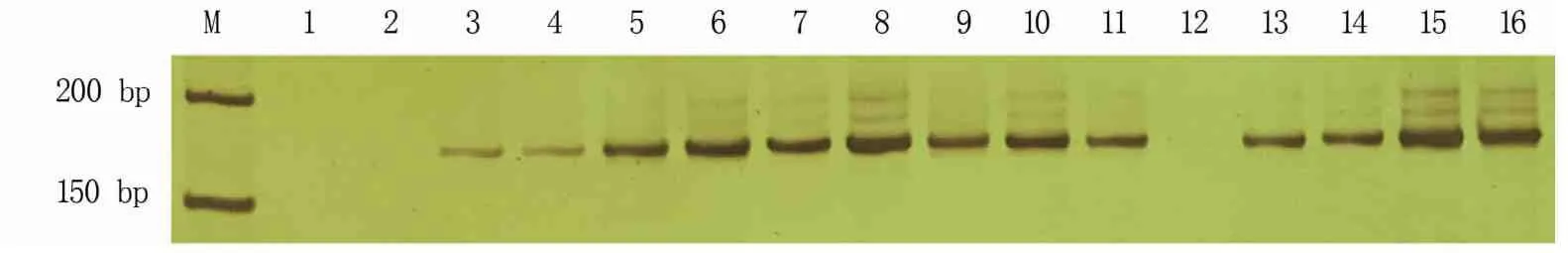
M为DL500 DNA Marker;1~16指正交试验设计的16个处理组合。
依据扩增出的特异条带清晰度并结合背景深浅进行打分,条带最清晰且无明显背景的打16分,以此类推,得到16个组合的分值依次为3、3、4、5、7、16、14、13、9、15、10、3、6、8、11、12,然后对同一因素相同水平下的分值平均值Xi(i=1,2,3,4,表示4个不同水平)以及各因素的极差R进行计算[14],结果(表4)显示,Mg2+用量对SSRPCR反应体系的影响最大,其次为引物和dNTPs,ExTaq 酶 和 模 板 DNA 影 响 较 小;组 合A2B2C1D4E1,即Mg2+用量1.0μL、引物用量0.5 μL、dNTPs用量0.2μL、ExTaq酶用量0.15μL、模板DNA用量15 ng时PCR扩增效果最好。
但由于ExTaq酶和模板DNA对SSR-PCR扩增结果的影响相对较小,结合单因素试验结果,综合考虑。最终确定含有Mg2+1.0μL、引物0.5 μL、dNTPs 0.2μL、ExTaq酶0.05μL、模板DNA 30 ng的PCR反应体系(10μL)最优。
2.4 优化反应体系的验证
利用8个国槐种质TA11、BZ26、DY17、ZB9、LY86、42、201、396和4对引物1302、1970、2541、3049对试验获得的最佳反应体系进行验证。结果(图8)显示,每对引物均扩增出清晰稳定的条带,其中引物1302和3049扩增出了多样性条带。表明上述试验确定的反应体系稳定、可靠,可用于国槐SSR标记开发及遗传多样性分析和指纹图谱构建等研究中。
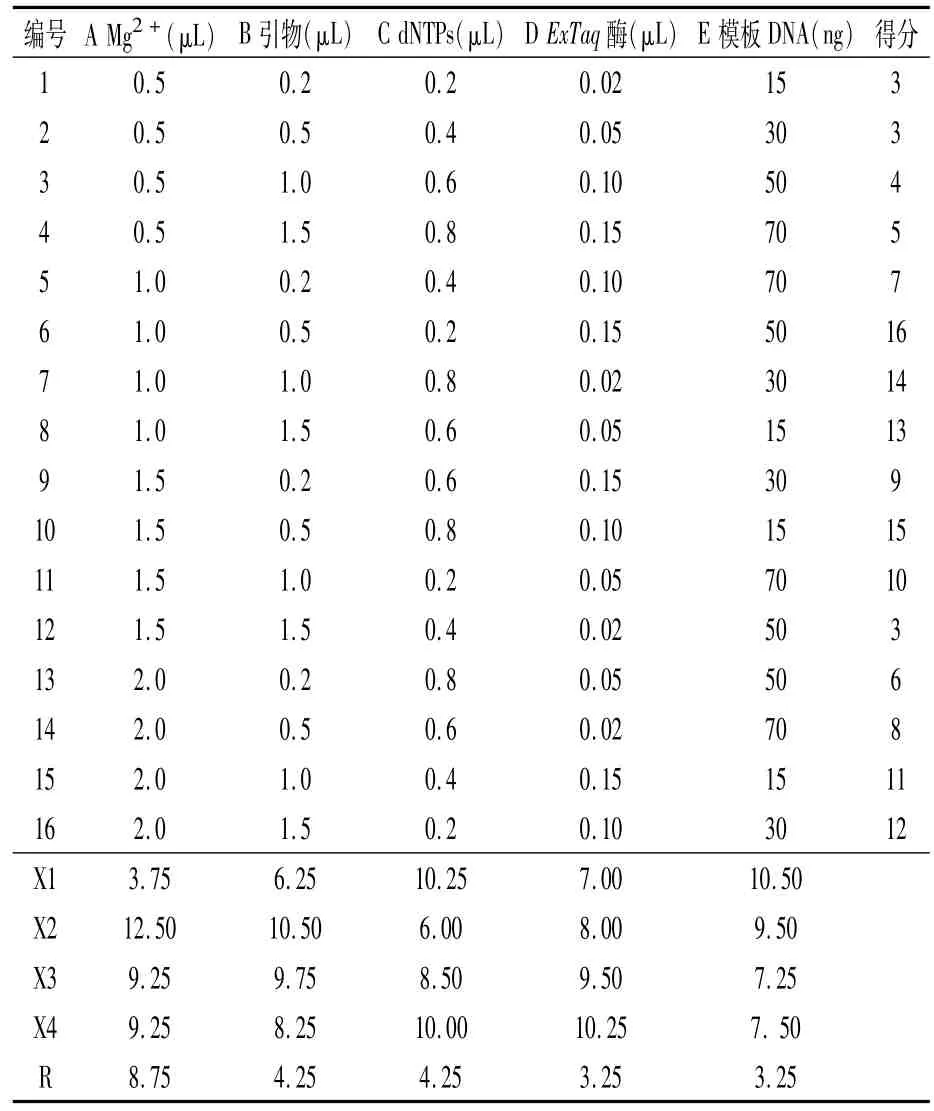
表4 国槐SSR-PCR反应体系优化的L16(45)正交试验设计及直观分析结果
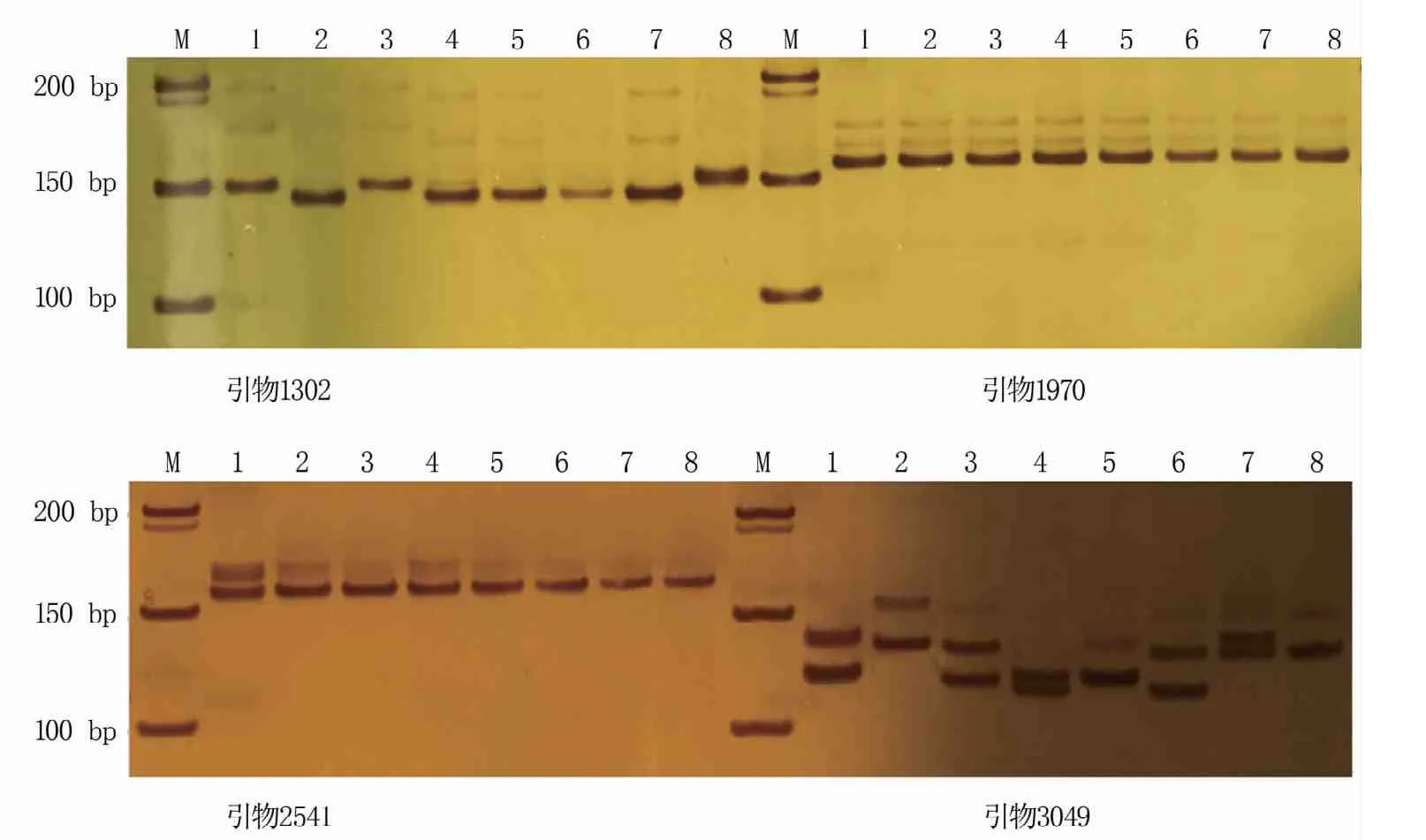
M指DL500 DNA Marker;1~8分别代表国槐种质TA11、BZ26、DY17、ZB9、LY86、42、201、396。
3 讨论与结论
PCR扩增是SSR分子标记实验的重要环节,可靠的PCR反应体系是开发和利用SSR标记的基础[15]。优化PCR反应体系,筛选最优因素组合,可以有效降低非特异性扩增,提高PCR扩增效率[16]。
单因素试验结合正交试验的方法能用较少的试验处理获得较好的试验结果,大大减少工作量。如利用该方法,宋伟栓[17]确立了适用于国槐SRAP的PCR反应体系,王健兵等[18]优化了白蜡SSR-PCR反应体系,赵克奇等[14]建立了适用于刺槐EST-SSR标记的PCR反应体系。
本研究利用单因素试验与正交试验相结合的方法对国槐SSR-PCR反应体系中的Mg2+、dNTPs、引物、ExTaq酶、模板DNA用量进行了优化,最终得到最佳反应体系(10μL):25 mmol/L Mg2+1.0μL,2.5 mmol/L dNTPs 0.2μL,10μmol/L上下游引物共0.5μL,5 U/μL ExTaq酶0.05μL,30 ng/μL DNA模板1.0μL,10×PCR Buffer 1.0μL,ddH2O 6.25μL。并选用4对引物、8个国槐种质对该PCR反应体系的扩增效果进行验证,结果表明在该反应体系下均能扩增出清晰、稳定的多态性条带。本试验结果为利用SSR标记深入研究国槐种质资源奠定了基础。
猜你喜欢
广西植物(2022年8期)2022-09-07
农业工程学报(2022年8期)2022-08-08
中国农学通报(2022年12期)2022-06-01
青年文学家(2022年12期)2022-05-18
自然灾害学报(2022年2期)2022-05-10
中国学校体育(2021年10期)2021-04-26
中学生物学(2019年7期)2019-10-17
诗潮(2017年12期)2018-01-08
科技资讯(2016年32期)2017-03-31
延河·绿色文学(2016年8期)2016-05-14
