
射血分数保留性心力衰竭潜在的分子机制及治疗靶点
2022-06-09 12:31石玉姣熊双刘春秋杨晨光董国菊刘剑刚
心血管病学进展 2022年5期
石玉姣 熊双 刘春秋 杨晨光 董国菊,2 刘剑刚,2
(1.中国中医科学院研究生院,北京 100700;2.中国中医科学院西苑医院心血管内科,北京 100091)
射血分数保留性心力衰竭(heart failure with preserved ejection fraction,HFpEF)是一种以心力衰竭(心衰)症状和体征,左室射血分数≥50%,及心脏结构改变(左心室重塑和/或左心房扩大)或舒张功能障碍为主要表现的临床综合征,约占心衰的50%[1]。其发病率和死亡率与射血分数降低性心力衰竭相似,并且随着人口老龄化、肥胖、糖尿病和高血压等合并症的持续流行而增加。由于HFpEF的病理生理机制复杂,导致目前治疗方法局限,且效果不佳[2]。目前,多种合并症诱发的全身慢性炎症被认为是驱动HFpEF发生的关键原因[3]。炎症可能通过抑制一氧化氮(nitric oxide,NO)-环磷酸鸟苷(cyclic guanosine monophosphate,cGMP)-蛋白激酶G(protein kinase G,PKG)信号通路;或通过激活转化生长因子β(transforming growth factor-β,TGF-β),刺激Smad依赖性和Smad非依赖性信号通路;或通过诱导冠状动脉微血管功能障碍(coronary microvascular dysfunction,CMD)的发生,最终引起心脏发生不良重构及舒张功能障碍等病理生理改变(见图1)。因此了解这种发病模式潜在的调控机制,有利于探索有效的治疗方法。现对HFpEF这种发病机制及相应治疗方法的研究现状进行综述。
1 分子机制
1.1 NO-cGMP-PKG信号通路
肥胖、糖尿病和高血压等多种合并症可诱发全身慢性炎症,促使血浆内白介素(interleukin,IL)-1、肿瘤坏死因子-α和C反应蛋白等炎症因子水平升高。炎症因子激活冠状动脉微血管内皮细胞,产生活性氧(超氧阴离子和过氧化氢等)和黏附分子(血管细胞黏附分子和E选择素)。超氧阴离子与NO反应形成过氧亚硝酸盐。过氧亚硝酸盐可氧化内皮型一氧化氮合酶(endothelial nitric oxide synthase,eNOS)的辅因子四氢生物蝶呤,引起eNOS的两个亚基解偶联形成单体,导致超氧阴离子生成增加,而NO生成减少且生物利用度下降。此外,活性氧能诱导可溶性鸟苷酸环化酶(soluble guanylyl cyclase,sGC)氧化或降解,导致sGC失活而不能再被NO激活。NO生物利用度下降及sGC失活导致cGMP的浓度下降,进而造成cGMP下游效应物PKG的活性降低。Franssen等[4]研究证实,HFpEF大鼠的冠状动脉微血管炎症标志物(血管细胞黏附分子和E选择素)上调,氧化应激增强[过氧化氢及单体eNOS明显上调,亚硝酸盐(NO2-)/硝酸盐(NO3-)浓度明显下调],cGMP浓度下降,PKG活性降低。并且在HFpEF患者左心室心肌活检试验中得到相似的结果。NO-cGMP-PKG信号下调,首先抑制cGMP及PKG介导的抗心肌细胞肥大机制,引起心肌细胞肥大,相关研究也证实了这一观点[5]。其次PKG活性降低导致肌联蛋白的N2 Bus位点低磷酸化,肌联蛋白作为一种跨越心肌肌节Z线和M线的巨大多肽,负责产生被动张力(F被动),其N2 Bus位点的低磷酸化导致左心室舒张期F被动增加,引起左心室心肌细胞僵硬及舒张功能障碍。Zhao等[6]研究证实,HFpEF模型小鼠较低的PKG活性与舒张期F被动增加相关。
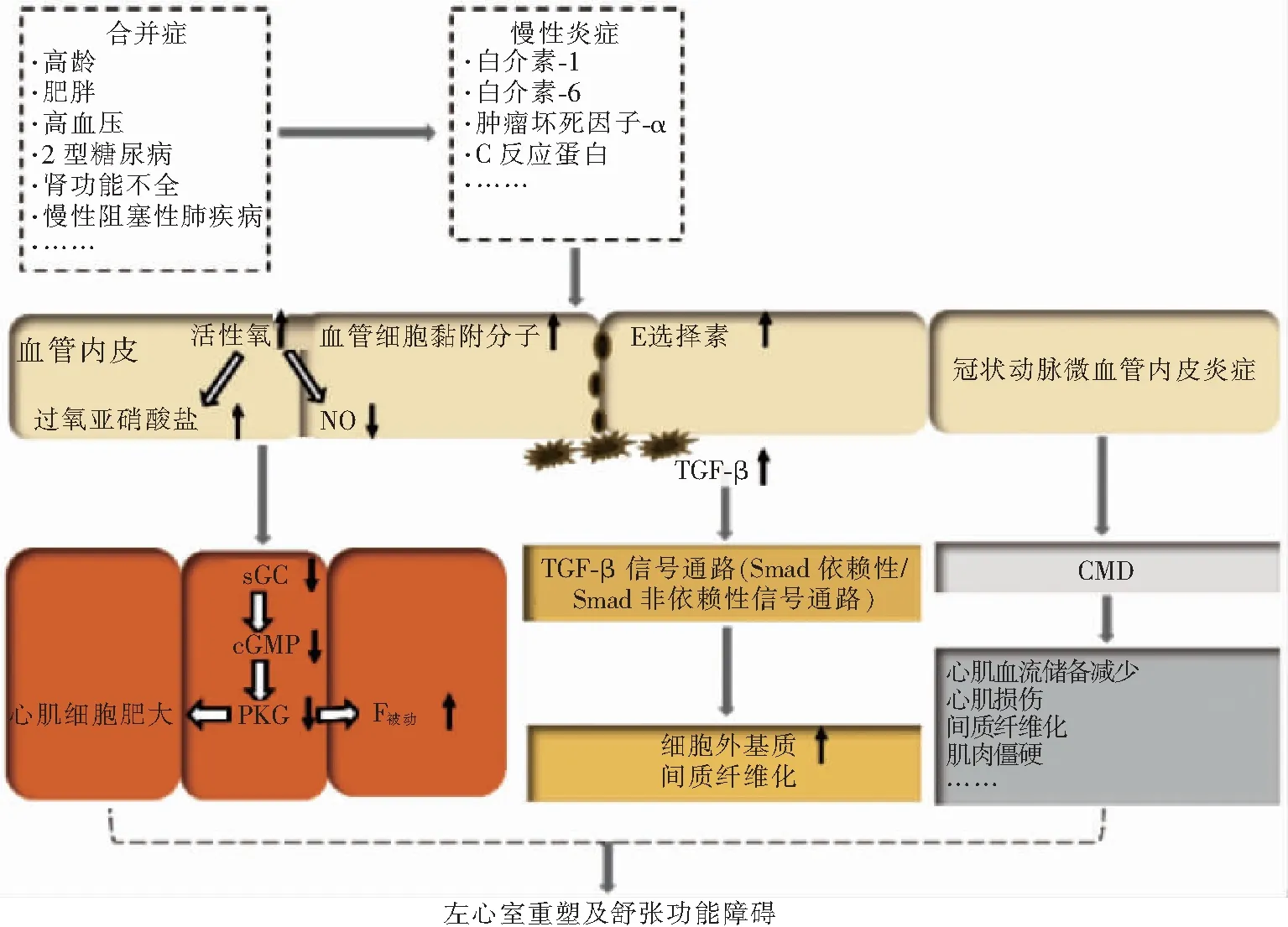
注:sGC为可溶性鸟苷酸环化酶。
1.2 TGF-β系统信号传导
黏附分子在慢性炎症的作用下,诱导冠状动脉微血管内单核细胞穿透血管壁,到达心脏间质,分化为巨噬细胞,并且在心脏间质驻留的巨噬细胞发生增殖,导致心肌间质的巨噬细胞增多,分泌的TGF-β增加。DeBerge等[7]研究证实,HFpEF患者心肌中巨噬细胞明显上调。TGF-β是TGF-β超家族的成员,参与细胞的生长、增殖、分化和凋亡等生理活动。TGF-β的3种亚型中TGF-β1在心肌纤维化中发挥主要作用。TGF-β在巨噬细胞中合成后,以无活性的状态储存在细胞间质中,在受到活性氧、血管活性激素(如血管紧张素Ⅱ)等刺激后被激活。活化的TGF-β与靶细胞上的跨膜受体结合,激活Smad依赖性和Smad非依赖性信号通路发挥下游效应。在Smad依赖性途径中,TGF-β结合TGF-βⅡ型受体(TβRⅡ),诱导TGF-βⅠ型受体(TβRI,也称ALK5)发生磷酸化。激活的ALK5特异性识别受体激活型Smads(Smad2和Smad3)并将其磷酸化,磷酸化的Smad2/3与共同介体Smads(Smad4)结合,形成功能性Smad2/3/4异源三聚体复合物,并转移到细胞核内,调节基因转录。而抑制性Smads(Smad6和Smad7)通过阻止Smad2/3的磷酸化及Smad2/3/4复合物的核易位抑制信号转导。TGF-β信号传导在诱导靶基因转录失调后,不仅导致成纤维细胞增殖,而且促使成纤维细胞分化为肌成纤维细胞,产生胶原蛋白。此外,活化的TGF-β不仅能抑制基质金属蛋白酶的表达,而且能诱导基质金属蛋白酶抑制剂的合成,将蛋白酶/抗蛋白酶平衡转向基质保留表型。心肌细胞外基质主要由Ⅰ型(约85%)和Ⅲ型胶原蛋白(6%~11%)组成。由于胶原蛋白的合成增加,降解减少,导致心肌细胞外基质沉积,心脏间质纤维化。有文献[8]报道,TGF-β-Smad2/3信号传导是心肌纤维化的主要参与者。而在TGF-β介导的Smad非依赖性途径中,与心肌纤维化发展密切相关的是3种丝裂原激活的蛋白激酶(mitogen-activated protein kinase,MAPK),包括细胞外信号调节激酶1/2(extracellular signal-regulated kinase 1/2,ERK1/2)、p38蛋白和c-Jun氨基末端激酶1/2(c-Jun N-terminal kinases 1/2,JNK1/2),以上信号传导均需转化生长因子β激活激酶1(TGF-β activated kinase-1,TAK1)的参与[9]。但TGF-β介导的Smad非依赖性途径涉及多种介质,其具体机制尚在探索中。Tan等[10]研究证实,HFpEF动物模型的TGF-β1、Smad2/3和ERK表达明显上调,并且与心肌细胞外基质沉积和心肌纤维化相关。而Roy等[11]发现,HFpEF患者心肌纤维化程度与舒张功能受损程度及不良事件(全因死亡率、心衰首次住院及再住院)明显相关。
1.3 CMD
近年来,全身炎症诱导的CMD在HFpEF发病中的重要作用逐渐受到重视。有研究[12-13]证实,在排除心外膜冠状动脉阻塞疾病后,HFpEF患者CMD患病率为72%~81%,并且CMD与心衰严重程度相关。然而迄今为止,CMD促使HFpEF发生的机制尚未明确,专家推测其可能通过多种分子通路,诱导心脏发生心肌血流储备减少、心肌损伤、局灶性和弥漫性纤维化、心脏脂肪变性、心肌僵硬、左心室功能障碍等病理改变[14]。Konerman等[15]研究证实,在排除冠状动脉阻塞疾病的情况下,HFpEF患者心肌血流储备减少,并且与左心室舒张功能障碍及顺应性降低相关。
2 治疗靶点
2.1 抑制炎症
IL-1阻滞剂通过竞争性结合IL-1受体,发挥抑制炎症效应。相关研究[16]显示,其代表药物anakinra可降低HFpEF患者血浆C反应蛋白和N末端脑钠肽前体水平,抑制炎症反应,并能改善左心室的舒张功能和运动能力。一项荟萃分析[17]显示,IL-1阻滞剂可降低心血管疾病复发的风险。因此笔者认为IL-1阻滞剂在HFpEF中的作用值得进一步探索。此外,Kolijn等[18]研究证实,恩格列净通过抑制炎症和氧化应激,刺激NO-sGC-cGMP信号传导,改善HFpEF小鼠左心室舒张功能;并且同期的体外试验也证实恩格列净通过抑制炎症及氧化应激,减轻HFpEF患者心肌细胞的僵硬度。EMPEROR-Preserved试验[19]则证实恩格列净能改善HFpEF患者的临床预后。
2.2 抑制肾素-血管紧张素-醛固酮系统
肾素-血管紧张素-醛固酮系统(renin-angiotensin-aldosterone system,RAAS)在HFpEF的炎症、氧化应激和左心室不良重塑等过程中发挥重要作用。既往认为,抑制RAAS可治疗HFpEF,然而几项关于RAAS抑制剂(坎地沙坦、厄贝沙坦、螺内酯和沙库巴曲/缬沙坦)治疗HFpEF的重要随机对照试验却显示阴性结果。但Kjeldsen等[20]考虑到HFpEF患者多伴有肥胖和高血压等多种合并症以及左心室收缩功能障碍,因此建议HFpEF患者常规使用RAAS抑制剂辅助治疗。而2021年2月美国食品药品监督管理局依据沙库巴曲/缬沙坦治疗HFpEF试验的次要结局,批准沙库巴曲/缬沙坦应用于HFpEF的适应证,并且随后的《2021 ESH高血压合并HFpEF的专家共识》也指出伴高血压的HFpEF患者应尽早使用沙库巴曲/缬沙坦[21]。近年,RAAS相关代谢产物血管紧张素转化酶2、血管紧张素1-7和血管紧张素1-9被证实可抵消经典RAAS的作用,在抗炎、抗氧化应激、改善内皮功能、抑制心肌细胞肥大和心肌纤维化等心血管相关疾病中发挥重要的作用[22]。因此可能是治疗HFpEF的潜在靶点,值得进一步研究。
2.3 调控NO-cGMP-PKG信号通路
2.3.1 增加NO
有机和无机NO3-均可通过增加NO含量,激活sGC-cGMP-PKG信号通路。然而,一项关于有机NO3-(单硝酸异山梨酯)治疗HFpEF的多中心随机对照试验[23]显示,有机NO3-不能改善患者的运动能力和生活质量,也不能降低N末端脑钠肽前体水平。并且长期使用有机NO3-会诱导药物耐受,以及存在低血压和头痛等副作用。因此无机NO3-作为HFpEF的潜在疗法逐渐引起研究者的重视。相关研究[24]表明,无机NO3-或NO2-可改善HFpEF患者运动能力及生活质量,但也有文献[25]报道相反的结果。然而考虑到以上关于无机NO3-或NO2-的试验样本量较小,且结果存在争议,因此后期可进行更大样本的研究以探索其疗效。
2.3.2 刺激sGC
sGC激动剂不依赖于NO直接激活sGC,增强下游cGMP信号。根据其起效方式可分为sGC刺激剂及sGC激活剂,其中sGC刺激剂能活化正常状态的sGC,并增强sGC对NO的敏感度;而sGC激活剂则能活化被氧化或辅基缺失的sGC,尤其在氧化应激或内皮功能障碍的情况下作用增强。但相关的多中心临床研究[26-27]显示,sGC刺激剂维利西呱及praliciguat不能改善HFpEF患者的生活质量及运动能力。而sGC激活剂在HFpEF中的运用尚处于临床前研究阶段。Kolijn等[28]发现sGC激活剂通过抑制炎症和氧化应激,改善sGC-cGMP-PKG信号传导,抑制心肌细胞肥大及降低舒张期F被动,进而改善高血压大鼠左心室的舒张功能。表明sGC激活剂是防治HFpEF的潜在选择,值得进一步探索。
2.3.3 抑制磷酸二酯酶
磷酸二酯酶Ⅴ型(phosphodiesterase type 5,PDE5)能分解cGMP,抵消cGMP-PKG信号传导,因此抑制PDE5是刺激cGMP-PKG信号的另一种方法。目前3种PDE5抑制剂(西地那非、他达拉非和伐地那非)主要用于勃起功能障碍和肺动脉高压,尚无一种药物被批准用于心衰。尽管临床前实验[29]证实西地那非可改善合并严重高血压和代谢综合征HFpEF大鼠的左心室僵硬和运动能力。但在既往相关临床研究[30]中,西地那非不能改善HFpEF患者运动能力及临床状态。并且相关研究[31]证实,西地那非可能会引起HFpEF患者线粒体功能障碍和内质网应激,导致与肾脏及心血管系统疾病相关的不良代谢产物增加,可见抑制PDE5可能不是防治HFpEF的理想靶点。
2.4 抑制TGF-β系统信号传导
抑制TGF-β1系统信号传导可减少胶原蛋白的产生或阻碍胶原蛋白的交联,从而防止心肌细胞外基质沉积,有可能成为防治心肌纤维化的主要方法。前期大量临床前试验证明了这一点(见表1)。然而TGF-β1信号传导具有多样性、多效性和交错性,限制了调控TGF-β1信号系统的治疗方法向具体临床运用的转化,因此目前尚无药物被批准用于改善HFpEF患者的心肌纤维化。然而2021年Lewis等[32]公布的一项Ⅱ期随机对照试验表明,TGF-β1抑制剂吡非尼酮可促使存在心肌纤维化的心衰患者(左室射血分数≥45%)的心肌细胞外体积减小和N末端脑钠肽前体水平下降。虽然该研究存在样本量较小及纳入样本年龄较大等缺陷,但其结果提示,吡非尼酮可改善HFpEF患者的心肌纤维化,值得进一步研究。
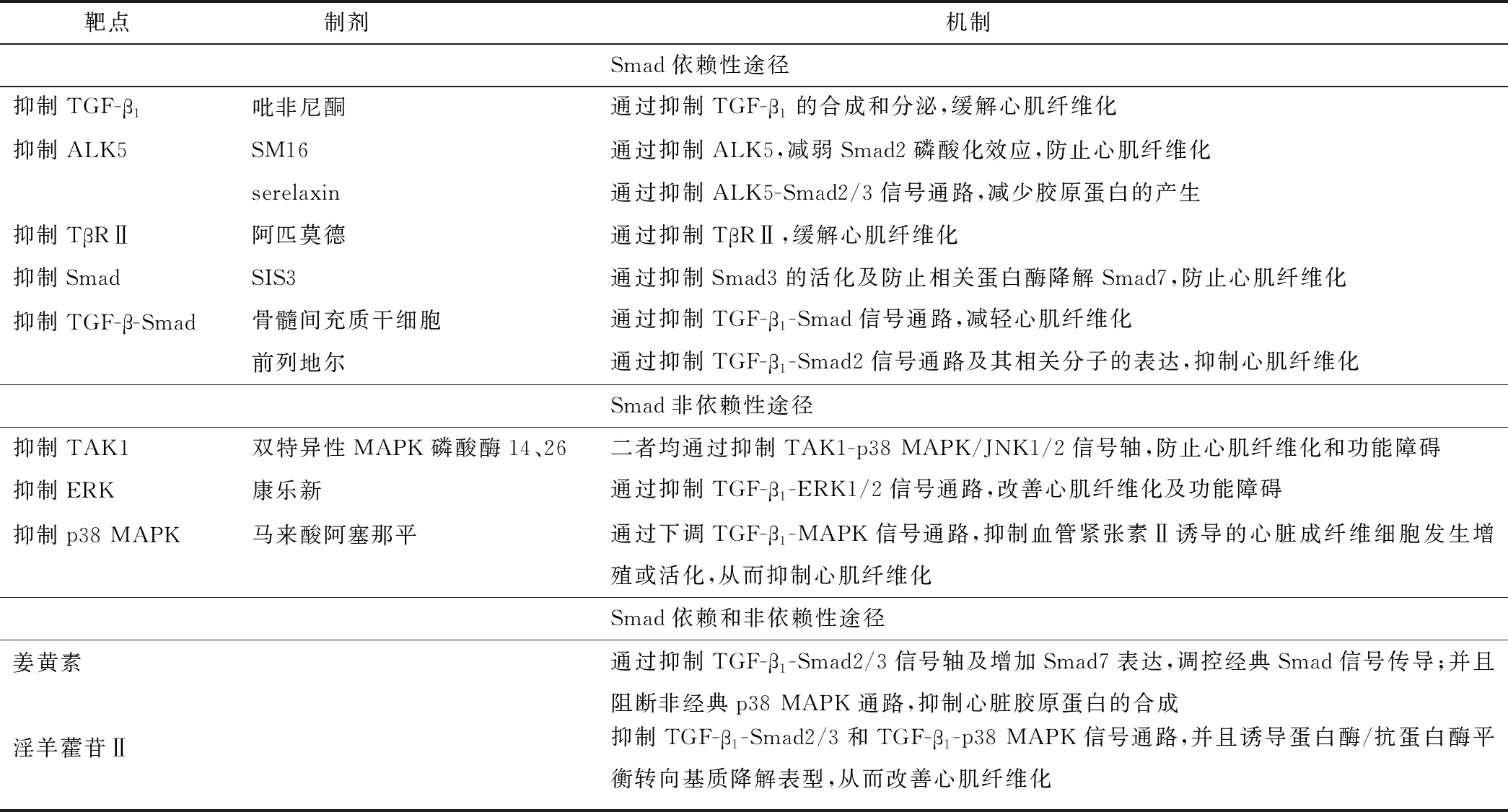
表1 抑制TGF-β1系统信号传导治疗心肌纤维化的相关靶点及制剂
2.5 改善CMD
改善CMD能抑制HFpEF的各种病理生理改变,然而CMD诱导HFpEF的发生机制尚未明确,致使针对性改善CMD治疗HFpEF的相关研究尚缺乏。尽管2020年ESC大会指出有机NO3-、RAAS抑制剂及他汀类药物等可能对改善CMD有益,但以上药物除了他汀类药物外,其他药物在HFpEF的治疗中均显示阴性结果[33]。既往的临床研究[34]证实,他汀类药物可降低HFpEF患者的死亡率,尤其对于无心外膜冠状动脉阻塞性疾病的HFpEF患者。可见他汀类药物可作为改善CMD的辅助药物,适当地运用于HFpEF的治疗中。
3 小结
综上所述,尽管HFpEF的病理生理机制复杂,但合并症诱导的全身炎症是驱动HFpEF患者发生左心室不良重塑及舒张功能障碍的关键原因。然而在这种疾病模式中,HFpEF的进展是以多种合并症为前提,以全身慢性炎症状态为驱动因素,并且涉及多种分子通路,致使当前针对性的治疗方法并未取得突破性进展。虽然抗炎、抗纤维化以及改善CMD似乎在一定程度上可延缓病程,但并不能改善患者的心衰结局。因此在未来HFpEF的治疗中,不仅要重视寻找能阻断炎症驱动HFpEF病理生理改变的治疗方法,也应重视对合并症进行治疗,通过加强运动训练,使用如沙库巴曲/缬沙坦、他汀类药物和恩格列净等目前证实能改善患者预后的药物进行辅助治疗。
猜你喜欢
现代临床医学(2022年4期)2022-09-29
中老年保健(2022年4期)2022-08-22
承德医学院学报(2022年2期)2022-05-23
昆明医科大学学报(2022年2期)2022-03-29
中西医结合心脑血管病杂志(2022年4期)2022-03-11
中西医结合心脑血管病杂志(2022年2期)2022-02-15
体育科学(2018年12期)2019-01-04
科学养生(2018年6期)2018-10-15
家庭百事通·健康一点通(2017年12期)2017-12-12
现代养生·上半月(2017年10期)2017-10-12
