
Tandem Mass Tag-based proteomics analysis reveals the vital role of inflammation in traumatic brain injury in a mouse model
2022-05-25 09:27JinQianDongQianQianGeShengHuaLuMengShiYangYuanZhuangBinZhangFeiNiuXiaoJianXuBaiYunLiu
中国神经再生研究(英文版) 2023年1期
Jin-Qian Dong ,Qian-Qian Ge ,Sheng-Hua Lu ,Meng-Shi Yang ,Yuan Zhuang ,Bin Zhang ,Fei Niu,Xiao-Jian Xu,,Bai-Yun Liu,,
Abstract Proteomics is a powerful tool that can be used to elucidate the underlying mechanisms of diseases and identify new biomarkers.Therefore,it may also be helpful for understanding the detailed pathological mechanism of traumatic brain injury (TBI).In this study,we performed Tandem Mass Tag-based quantitative analysis of cortical proteome profiles in a mouse model of TBI.Our results showed that there were 302 differentially expressed proteins in TBI mice compared with normal mice 7 days after injury.Gene Ontology and Kyoto Encyclopedia of Genes and Genomes pathway analyses showed that these differentially expressed proteins were predominantly involved in inflammatory responses,including complement and coagulation cascades,as well as chemokine signaling pathways.Subsequent transcription factor analysis revealed that the inflammation-related transcription factors NF-κB1,RelA,IRF1,STAT1,and Spi1 play pivotal roles in the secondary injury that occurs after TBI,which further corroborates the functional enrichment for inflammatory factors.Our results suggest that inflammation-related proteins and inflammatory responses are promising targets for the treatment of TBI.
Key Words:bioinformatics;complement cascade;mass spectrometry;neuroinflammation;proteomics;secondary injury;subacute phase;tandem mass tag;transcription factor;traumatic brain injury
Introduction
Traumatic brain injury (TBI) is a leading cause of mortality and disability across all age groups and imposes a tremendous burden on families and society(Maas et al.,2017).Increasing evidence shows that TBI is a chronic disease process (Masel and DeWitt,2010;Gilmore et al.,2020) that is associated with neurodegenerative diseases (Xu et al.,2021) and mental health disorders(Stein et al.,2019).However,to date,no effective treatments are available to promote functional recovery (Ghiam et al.,2021).Therefore,gaining a better understanding of the pathophysiology of TBI is critical to breaking the bottleneck for the treatment of patients with TBI (Dixon,2017;Thapa et al.,2021).
Omics-based technologies,including genomics,transcriptomics,proteomics,and metabolomics (Samia Hassan,2019),have revolutionized biomedical research and provided a more holistic perspective of specific biological functions.The aim of omics-based approaches is to collectively identify,characterize,and quantify all phenotype-associated biomolecular changes(Vailati-Riboni et al.,2017;Abu Hamdeh et al.,2021).Proteins are the ultimate executors of physiological processes (Tang et al.,2019) and key indicators of disease states (Williams et al.,2019).Therefore,interrogating changes in protein dynamics,which is essentially the scope of proteomics,provides crucial information for disease-specific diagnosis and treatment(Krzyszczyk et al.,2018).Proteomics,a discipline that has arisen in the postgenomic era,comprises proteome analysis and large-scale systematic analysis of protein expression (Pandey and Mann,2000;Hristova and Chan,2019).An individual’s genome is unique and constant,whereas the proteome is diverse and dynamic,which makes proteomics a powerful tool for elucidating the underlying mechanisms of diseases and identifying novel biomarkers.
Due to the complexity of the brain and the heterogeneity of TBI-related pathogenesis (Jassam et al.,2017;Yang et al.,2021),the detailed pathological mechanisms underlying TBI are not completely known (Qin et al.,2021).The TBI field could benefit from the use of proteomics analyses,given the diversity of primary insults and secondary injury-associated processes (Sarkis et al.,2017).Proteomics studies of TBI have involved a variety of species,animal models,tissue samples,time points,and approaches (Xu et al.,2016;Ojo et al.,2018;Pham et al.,2021;Wang et al.,2021b;Zhang et al.,2021).Moreover,technical development in proteomics,such as the tandem mass tag (TMT) labeling approach,can provide novel methods for TBI proteomic investigation (Thompson et al.,2003;Zhang et al.,2014).
The subacute phase,which is often defined as 7 days post-injury,is a turning point in the pathophysiology of TBI (Kaindl et al.,2007;Guglielmetti et al.,2017;Robinson et al.,2017).In the present work,we took advantage of TMTbased proteomics and a mouse model of controlled cortical impact (CCI) to identify critical proteins and biological processes involved in the subacute phase of TBI.
Methods
Animals
Twenty specific pathogen-free male C57BL/6 mice (2 months old) weighing 24–26 g were supplied by Beijing Vital River Laboratory Animal Technology Co.Ltd.(Beijing,China;license No.SCXK (Jing) 2016-0006).All mice were maintained in groups of five mice per cage in a barrier housing facility with a 12/12-hour light/dark cycle.Laboratory rodent chow and tap water were providedad libitum
.All animals in this study were acclimated for at least 2 weeks before any procedures were performed.The mice were randomized into either the CCI (n
=10) or the control group (anesthesia only,n
=10).The study was approved by the Animal Welfare Ethics Committee of Beijing Neurosurgical Institute,Capital Medical University on March 24,2020(approval No.201904011).All experiments were designed and reported according to the Animal Research:Reporting ofIn Vivo
Experiments (ARRIVE)guidelines (Percie du Sert et al.,2020).During the surgical procedure,a small animal anesthesia machine (RWD Life Science Co.,Shenzhen,China) was used for induction or maintenance of general anesthesia via inhalation anesthesia with isoflurane (RWD Life Science Co.) at concentrations of 2% or 1.5%,respectively.The flow chart was shown inFigure 1
.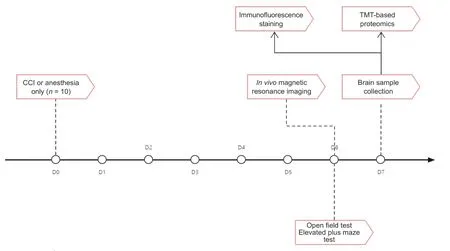
Figure 1 | Study flow chart.
CCI model
TBI was induced in mice utilizing a CCI model,as previously described (Smith et al.,1995).To make the mice more comfortable and reduce the risks associated with anesthesia,a temperature maintenance device (RWD Life Science Co.) was used when the mice were under anesthesia.Following loss of consciousness,the head was secured in a stereotaxic instrument (RWD Life Science Co.) that held the head firmly without damaging the skull.After depilation,iodophors were used to sterilize the surgical site.The following procedures were performed using aseptic techniques.A midline scalp incision was performed and the muscles were retracted to expose the calvarium.Then,a 5 mm-diameter craniotomy was performed over the parietal cortex,2 mm to the right of the midline,between lambda and bregma (Wu et al.,2021),using a dental drill,while keeping the dura undamaged.The PCI3000 PinPoint Precision Cortical Impactor (Hatteras Instruments,Cary,NC,USA)with a 3 mm-diameter impactor tip at a velocity of 3 m/s,depth of 1.5 mm,and dwelling time of 20 ms was used to induce moderate TBI.Finally,the skull defect was repaired with bone wax,and the scalp incision was closed with stitches.
In vivo magnetic resonance imaging
At 6 days post-injury,in vivo
magnetic resonance imaging (MRI) was used to confirm the radiological abnormalities characteristic of the subacute stage of TBI.The mice were scanned using a 7.0 Tesla small-bore animal scanner (Bruker Biospec,Ettlingen,Germany).Mice were anesthetized with 2% isoflurane in ambient air,then fastened to an adapter to immobilize the heads.During the imaging,anesthesia was maintained using isoflurane via a nasal catheter,and respiration was monitored using the monitoring system that the scanner is equipped with (Bruker Biospec).T2-weighted structural images were obtained using axial,2D,T2-TurboRARE sequences.The imaging parameters were as follows:repetition time=2795 ms,echo time=35 ms,echo spacing=11.667 ms,rare factor=8,field of view=22 mm × 22 mm,and slice thickness=0.5 mm.MRI analysis
MRI is a sensitive and noninvasive tool that can be used to detect lesions in the brain.Brain lesions were defined as focal regions of abnormally hypoor hyperintense signals in the white or gray matter.Brain lesion volume was measured on the T2-weighted image (T2WI) using a semiautomatic segmentation method.ITK-SNAP (version 3.8.0) (Yushkevich et al.,2006) was used to trace and calculate regions of interest on 25 consecutive axial MRI T2-weighted structural images.First,the structure of interest was defined.Next,the multiple structural images were transformed into a speed image with the voxels inside the lesion and non-lesion having positive and negative speed values,respectively.Then,seeds were placed inside the structure of interest.Last,seeds were expanding to fill the structure of interest and the segmentation was completed.The ITK-SNAP automatically calculates the volume after the segmentation of the structure.All of the MRI images were acquired and analyzed by a researcher who was blinded to the experimental injury conditions.
Behavioral assessment
Small rodents,including mice,have an innate aversion to unfamiliar open fields and intense light (Choleris et al.,2001).Thus,the elevated plus maze(EPM) and open field test (OFT) were used to evaluate anxiety-like behavior in the mice.All behavioral assessments were performed at 6 days postinjury.One hour before the experiment,the mice were transferred into the behavioral testing room for acclimation.The apparatuses were cleaned with 70% ethanol before each test.The tests were performed as described previously (Chen et al.,2021).An overhead night vision camera was used to record the activities of the mice.A blinded investigator carried out all behavioral assessments and data analyses.
Elevated plus maze
The EPM includes a central platform (10 cm × 10 cm),a pair of open arms(50 cm × 10 cm),and a pair of closed arms (50 cm × 10 cm × 10 cm).The apparatus was elevated 50 cm above the ground.The mouse was gently placed in the center of the maze,facing a closed arm.The free and uninterrupted activity of the mouse was recorded for 5 minutes.The time each mouse spent in the open arms and the number of times it entered the open arms were quantified using the SMART video tracking system (SMART v3.0,Panlab,Spain).
Open field test
An opaque plastic container (24 cm × 24 cm × 50 cm) and a lid with a hole in the center to accommodate the night vision camera were used for the OFT.For each trial,a mouse was gently grasped and placed in the middle of the bottom of the container.The mouse was allowed to move freely and uninterruptedly for 5 minutes and was monitored by the camera.The total distance traveled and the time spent in the inner area (8 cm × 8 cm) of the open field were measured using the SMART video tracking system (SMART v3.0).
Sample collection
Brain samples were collected at 7 days post-injury.For TMT-based proteomics,mice were anesthetized and then perfused with cold physiological saline via the left ventricle.The mouse was decapitated after confirmed death,and the entire brain was isolated.Approximately 2 mm of tissue representing the ipsilateral cortices around the injury site was dissected from the CCI mouse brains,and the corresponding part of the control mouse brains were dissected,as described previously (Wang et al.,2021b).The brain samples were placed in cryogenic storage tubes and flash-frozen using liquid nitrogen,after which they were stored at–80°C until further processing.
For immunofluorescence imaging,mice were euthanized and then perfused with cold physiological saline and 4% paraformaldehyde.Next,the cortices were extracted and fixed in 4% paraformaldehyde overnight at 4°C.The next day,the samples were immersed in 30% sucrose in phosphate-buffered saline(PBS) until they sank,at which point they were cut into 20 µm-thick coronal sections.
Protein extraction,digestion,and TMT labeling
The cortices were homogenized in SDT lysis buffer (2% sodium dodecyl sulfate,100 mM dithiothreitol,100 mM Tris-HCl,pH 7.6) twice at 6.0 m/s for 30 seconds each time using a FastPrep-24 Classic homogenizer (MP Biomedicals,Irvine,CA,USA).After sonication,the homogenate was incubated in boiling water for 15 minutes and centrifuged at 14,000 ×g
for 40 minutes.The amount of protein in the filtered supernatant was quantified using a bicinchoninic acid protein assay kit (Beyotime,Shanghai,China).Then,20 µg of protein from each sample was individually mixed with 6×loading buffer and boiled for 5 minutes.The boiled proteins were separated on a 12.5% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gel and subjected to Coomassie Blue R-250 staining for visualization.Protein digestion was performed using the filter-aided sample preparation method,as previously described (Wiśniewski et al.,2009;Wang et al.,2021a).The samples were labeled as TBI1-126,TBI2-127,TBI3-128,CON1-129,CON2-130,and CON3-131,with three biological replicates per group,using a TMT 6 plex Isobaric Mass Tag Labeling kit (Thermo Fisher Scientific,Waltham,MA,USA).Afterward,the TMT-labeled peptides were fractionated by reversephase chromatography using a 1260 Infinity II High Performance Liquid Chromatography apparatus (Agilent Technologies,Santa Clara,CA,USA),as described earlier (Wang et al.,2021a).Mass spectrometry analysis
All fractions were injected for nano-liquid chromatography-tandem mass spectrometry analysis.The mixture of peptides was loaded onto a C18 reverse-phase analytical column (Thermo Fisher Scientific) in buffer A (0.1%formic acid) and diluted with a linear gradient of buffer B (0.1% formic acid and 80% acetonitrile) at a flow velocity of 300 nL/min.
A Q Exactive Plus mass spectrometer was used,coupled to an Easy nLC system (Thermo Fisher Scientific) and manipulated in positive ion mode.Mass spectrometry (MS) data were acquired via a data-dependent top 10 method that dynamically chose the most abundant precursor ions from the survey scan for higher-energy collisional dissociation fragmentation.The parameters of the survey scans were as follows:resolution=70,000,AGC target=3e6,maximum injection time=50 ms.The parameters of the MS2 scans were as follows:resolution=17,500,AGC target=3e6,maximum injection time=45 ms,isolation width=2 m/z.Only ions with a minimum intensity of 2e3 and a charge state between 2 and 6 were chosen for fragmentation.
Raw data were processed using the MASCOT engine 2.6 (Matrix Science,London,UK) embedded in the Proteome Discoverer 2.2 software program(Thermo Fisher Scientific).Protein sequences for Mus musculus were collected from the UniProt database.The principal parameters were set as follows:max missed cleavages=2;precursor mass tolerance=± 10 ppm;fragment mass tolerance=0.05 Da;and peptide false discovery rate ≤ 0.01.Differentially expressed proteins (DEPs) were defined as those exhibiting a fold change >1.2 for upregulation and <0.83 for downregulation atP
<0.05,in accordance with previous studies (Casey et al.,2017;Song et al.,2019).Bioinformatics analysis
Gene Ontology (GO) was used to annotate the functions of the DEPs in three categories,namely,biological process,cellular component,and molecular function (Ashburner et al.,2000).Kyoto Encyclopedia of Genes and Genomes(KEGG) pathway analysis was applied to identify signal transduction pathways and biochemical metabolic pathways related to the DEPs (Kanehisa and Goto,2000).The open-access platform Metascape (https://metascape.org)was used to perform the GO and KEGG analyses.The Benjamini-Hochberg procedure was performed to compute false discovery rates to correct for multiple comparisons.A false discovery rate of less than 0.05 was considered significant enrichment.The DEP volcano plot and heatmap were graphed using R software (https://www.r-project.org/).The volcano plot was generated based on log2(fold change) at thex
-axis and -log10(p
-value) at they
-axis.The Animal Transcription Factor DataBase (AnimalTFDB,http://bioinfo.life.hust.edu.cn/AnimalTFDB/#!/) was used to annotate transcription factors(TFs) (Hu et al.,2019).Differentially expressed genes (DEGs) and TFs after TBI were imported into the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING;https://string-db.org) database to identify protein-protein interactions (PPIs).The open-source software Cytoscape (https://cytoscape.org) was used to visualize the PPI network (Shannon et al.,2003).The cytoHubba plugin in Cytoscape was used to predict and discover hub genes and hub TFs with the highest node degrees via the Degree method,which is a local-based topological analysis method provided by cytoHubba (Chin et al.,2014).TF regulatory networks were visualized using Cytoscape software.For all TMT-based proteomics analyses,the investigator was blinded to the raw data.
Immunofluorescence staining
Immunofluorescence staining was performed on the sections taken from CCI and control mice as described previously (Henry et al.,2020).Brain tissue located 0.5 mm to–2.5 mm from the bregma based on stereotaxic coordinates was dissected out and sectioned.After washing three times in PBS,the sections were blocked in goat serum (Zhongshan Goldenbridge Biotechnology,Beijing,China) for 1 hour at room temperature,and then incubated overnight at 4°C in the primary antibodies,including rabbit anti-C3 (1:300,Proteintech,Rosemont,IL,USA,Cat# 21337-1-AP,RRID:AB_2878843) and rabbit anti-Iba1 (official symbol:allograft inflammatory factor 1,Aif1;1:500,Wako Chemicals,Richmond,VA,USA,Cat# 019-19741,RRID:AB_839504).After 24 hours,the sections were washed with PBS threetimes and incubated in Cy3-conjugated goat anti-rabbit IgG (1:300,Abcam,Cambridge,UK,Cat# ab97080,RRID:AB_10679808) at room temperature for 1 hour to visualize the target antigens.Finally,the sections were washed with PBS three times,counterstained with 4,6-diamidino-2-phenylindole(Thermo Fisher Scientific),and mounted with coverslips.The sections were then imaged using a ZEISS/LSM 880 confocal laser scanning microscope (Carl Zeiss AG,Oberkochen,Germany).ImageJ software (National Institutes of Health,Bethesda,MD USA) (Schneider et al.,2012) was used to quantify the immunofluorescence signal intensities by a blinded investigator.
Statistical analysis
The statistics program GraphPad Prism 8.0.2 (GraphPad Software,San Diego,CA,USA,www.graphpad.com) was used for statistical analyses and graphical representations.The variance in lesion volumes on MRI was measured using relative standard deviation.For the behavioral assessment and immunofluorescence imaging,differences were analyzed by unpairedt
-test to compare the control and CCI groups.Differences in the average expression levels of a given protein between two groups were assessed by independent two-samplet
-test.Data are presented as mean ± standard error of the mean (SEM) and coefficient of variation.For all comparisons,the statistical significance was set as aP
-value of less than 0.05.Results
Brain lesions on T2-weighted MRI after CCI
Brain abnormalities were investigated by T2WI at 6 days post-injury.As illustrated inFigure 2A
,abnormal hypo-or hyperintensity was detected at the injury site on T2WI.Compared with controls,CCI caused brain edema and structural abnormalities in the ipsilateral hemisphere,including the cortex,hippocampus,corpus callosum,and lateral ventricles.No evidence of pronounced midline shift was found on T2WI in CCI mice.There was minimal variability in the lesion volumes of the three CCI mice used for proteomic analysis (8.637 ± 0.219 mm,coefficient of variation=4.381%;Figure 2B
),indicating consistency among the biological replicates and the reliability of the proteomics data.Behavioral changes after CCI
The anxiety levels of mice are inversely proportional to the time spent in the open arms of the EPM or the inner area during the OFT (Shavit-Stein et al.,2021).CCI animals spent significantly less time in the open arms of the EPM compared with the control group (39.31± 10.13 secondsvs
.80.15 ± 15.32 seconds,P
=0.0397;Figure 2C
).The difference between the two groups in terms of the number of times the mice entered the open arms fell just short of statistical significance (7.000 ± 0.8498vs
.9.800 ± 1.685,P
=0.1213).In the OFT,CCI mice spent significantly less time in the inner area compared with control mice (9.68 ± 1.68%vs.
20.26 ± 4.65%,P
=0.0277;Figure 2D
).However,the total traveled distance in the OFT did not differ between the groups (1148 ± 135.9 cmvs
.1191 ± 150.3 cm,P
=0.8391).Altogether,these results indicated that TBI in the subacute stage increased anxietylike behavior,as demonstrated by a decreased risk-taking behavior in an unfamiliar environment.Quantitative proteomic analysis of cortical proteome profiles after CCI
To gain a detailed understanding of the molecular changes that occur during the subacute stage of TBI,the proteomic profiles in six mouse cortex samples from the CCI and control groups were analyzed using TMT 6-plex tagging and LC-MS/MS.Principal component analysis was performed to estimate variations between and within groups.The principal component analysis plot demonstrated a clear distinction between groups and consistency within each group (Figure 3A
).We identified and quantified a total of 5738 proteins in cortex tissues 7 days post-TBI via large-scale proteomics analysis (Additional Table 1
).Twenty of these proteins were upregulated,and 100 were downregulated.A volcano plot of the DEPs is shown inFigure 3B
.A heatmap of the DEPs showed distinct protein profiles for the CCI and control samples (Figure 3C
).Hierarchical clustering analysis demonstrated a trend toward increased or decreased protein expression following subacute TBI.These results demonstrate that protein expression profiles are significantly altered in the subacute phase(7 days) following CCI.Additionally,the majority of the DEPs appeared consistently within all members of a single group,which indicated good reproducibility.GO functional classification of DEPs after CCI
The enrichment analysis was performed to determine the function of the DEPs according to the three aspects mentioned above.The 20 most significantly enriched GO terms,consisting of two biological processes,five molecular functions,and thirteen cellular components,are shown inFigure 4A
.Cell adhesion and biological adhesion were the most overrepresented biological processes.The significantly enriched molecular functions comprised cell adhesion molecule binding,identical protein binding,actin-binding,integrin binding,and volume-sensitive anion channel activity.As for the cellular component annotation,the significantly enriched terms included adherens junction,anchoring junction,cell junction,focal adhesion,cell-substrate adherens junction,cell-substrate junction,and more.GO enrichment analysis of the DEPs illustrated that these proteins are related to the immune system and inflammation-related processes.KEGG pathway analysis of DEPs after CCI
A pathway represents the sequence of chemical reactions in the cell that causes a particular biological effect (Schmidt et al.,2014).KEGG pathway analysis demonstrated that the significantly enriched pathways in the CCI group were mainly involved in inflammation (Figure 4B
).Among the identified KEGG pathways,Fc gamma R-mediated phagocytosis exhibited the highest statistical significance and the largest number of genes,includingASAP2
,FCGR2
,FCGR1
,RAC2
,andLYN
.Additionally,inflammation-related pathways such as complement and coagulation cascades,leukocyte transendothelial migration,antigen processing and presentation,and the B cell receptor signaling pathway were significantly enriched and share proteins with altered expression levels such asRAC2
,FCGR2
,andLYN
.Other noticeably enriched pathways included lysosome,platelet activation,cell adhesion molecules,chemokine signaling pathway,and glutamatergic synapse.Taken together,KEGG pathway analysis of the DEPs revealed that these proteins are mainly involved in inflammatory signaling pathways,which was consistent with the GO annotation results.PPI network construction and identification of hub genes based on DEPs after CCI
PPI networks are interaction networks that represent the high connectivity and complexity of hub proteins.PPI network analysis of the DEPs showed direct and indirect interconnections among 302 DEPs.Based on the generated PPI network,the ten most highly connected hub genes,which are the significant nodes in PPI networks and represent DEGs with a large number of associations with other DEGs (Chen et al.,2020a),were identified (Figure
4C
).Among these hub genes,Lyz2
exhibited the most significant difference between the CCI and control groups.GO and KEGG analyses demonstrated that the hub genes,such asItgb2
,Icam1
,Fermt3
,Lcp1
,Aif1
,andPtprc
,were primarily associated with inflammatory processes and complement systems.Immunofluorescence analysis further confirmed the significant upregulation ofAif1
in the CCI group compared with the control group (Figure 4D
).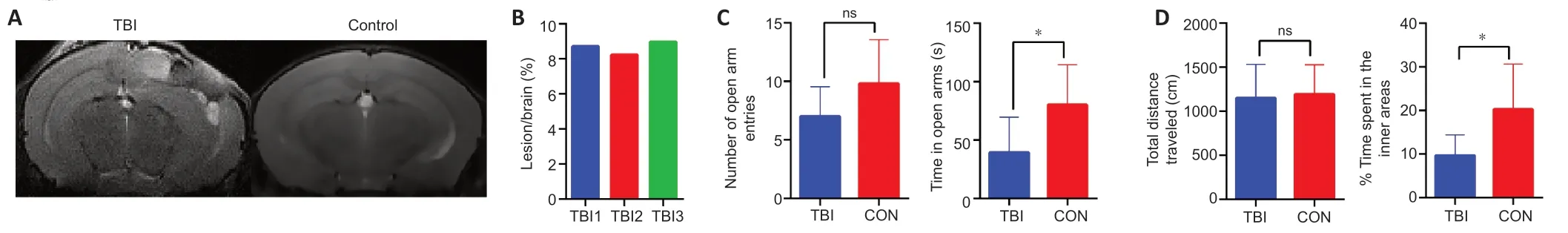
Figure 2 | Neuroimaging and neurobehavioral alterations at 6 days post-TBI.
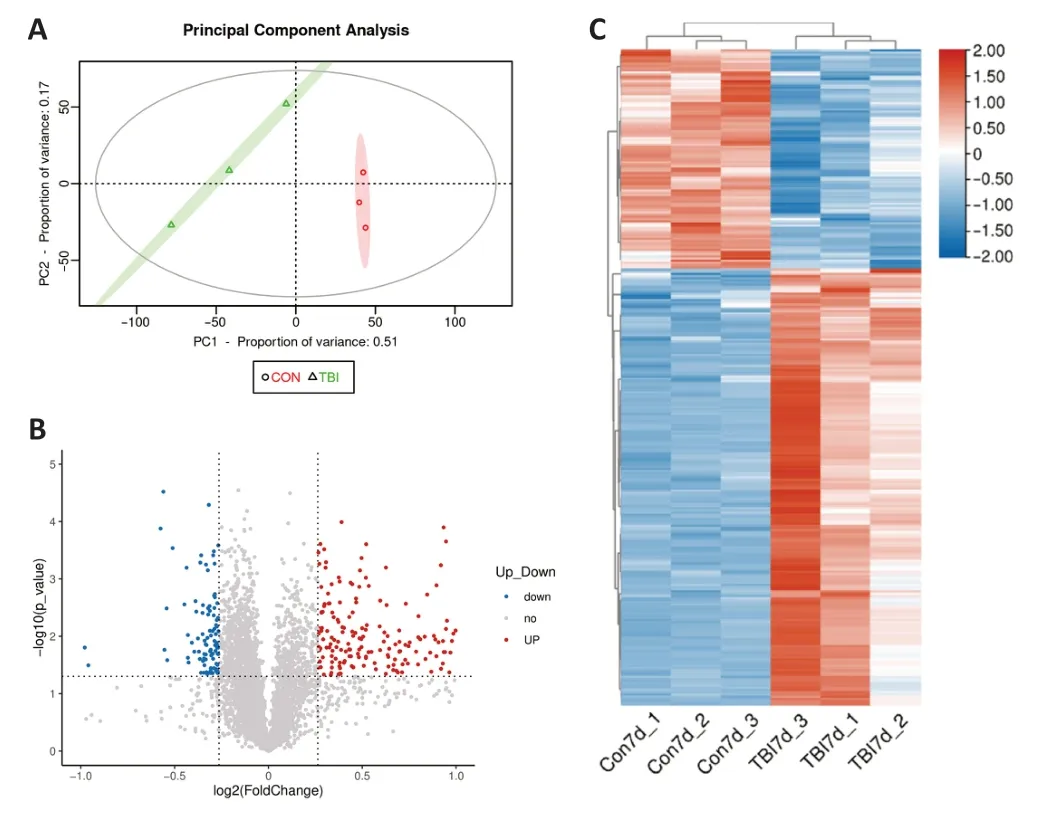
Figure 3 | Analysis of cortical DEPs in TBI and control mice.
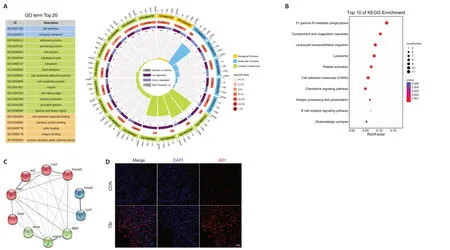
Figure 4 | Gene Ontology (GO),Kyoto Encyclopedia of Genes and Genomes(KEGG),and protein-protein interaction (PPI) analysis of cortical DEPs between the TBI and control mice.
TF regulatory network analysis
TFs control the initiation of gene transcription,regulate gene expression,and play an essential role in almost all biological processes (Lambert et al.,2018;Mitsis et al.,2020).Therefore,TBI-related alterations in TF expression were investigated to gain better insight into the pathophysiologic mechanism of TBI.The nine most connected hub TFs were identified from the PPI networks:NF-κB1,RelA,STAT1,IRF1,CEBPA,Spi1,E2F1,SP1,and NFYA.Next,genes upregulated and downregulated by the hub TFs were predicted and used to construct a TF regulatory network that included 46 edges,nine TFs,and 27 target genes (Figure 5
).Among the identified hub TFs,NF-κB1,RelA,STAT1,IRF1,and Spi1 played pivotal roles in the inflammatory response.Complement C3 is upregulated following TBI,as detected by immunofluorescence
The functional analyses described above suggested that the complement cascade may exert a detrimental effect in the context of TBI.To confirm this finding,the expression of the hub protein complement C3,which is a central molecule in the complement cascade and the inflammatory response (Ricklin et al.,2016),was investigated by immunofluorescence.There was a highly statistically significant increase in total C3 protein immunopositivity in cortices from mice 7 days post-TBI compared with control mice (P
<0.001;Figure 6
),which verified the bioinformatics-based functional analysis.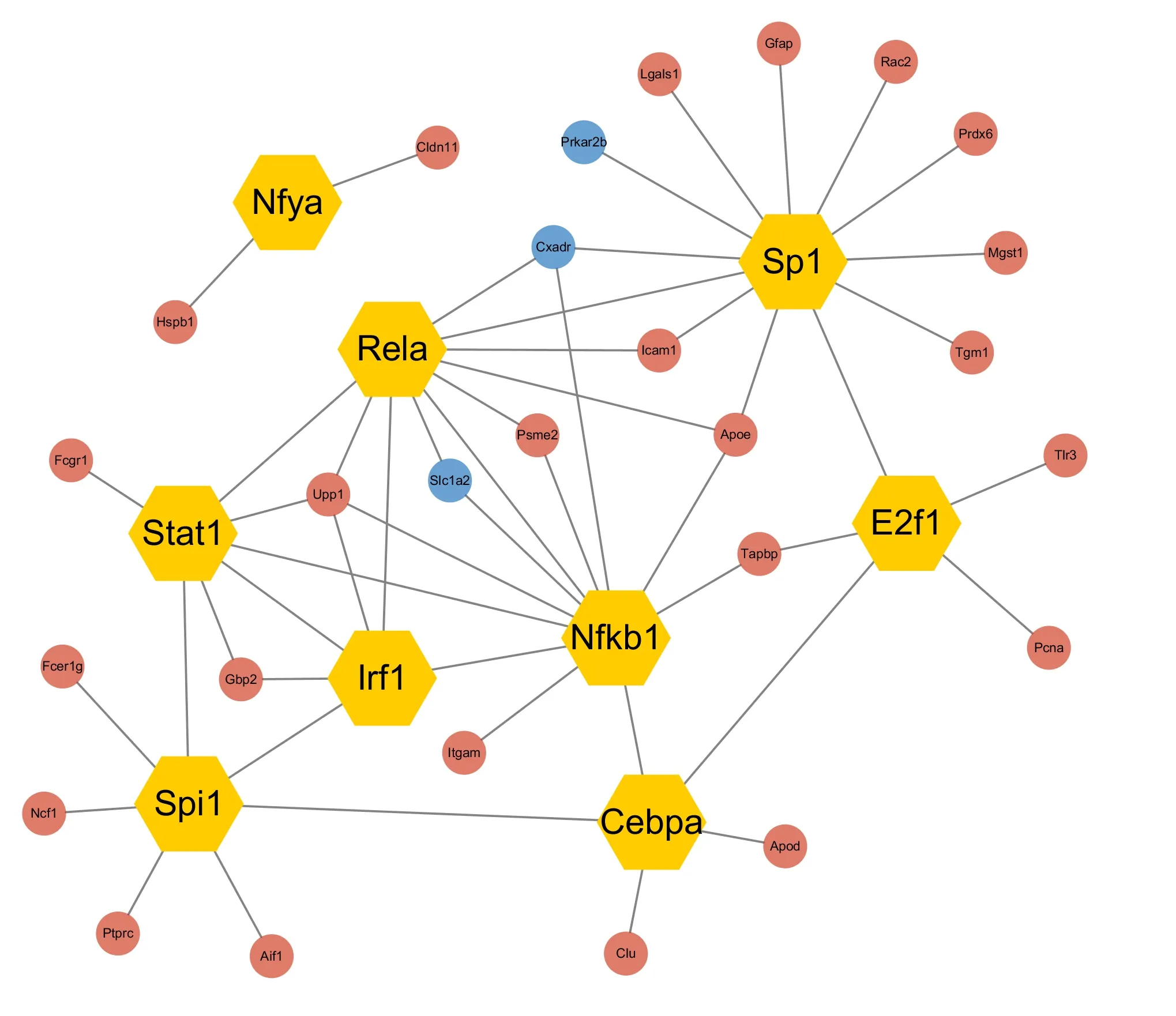
Figure 5 | Regulatory network analysis of transcription factors that were differentially expressed in TBI and control mice.
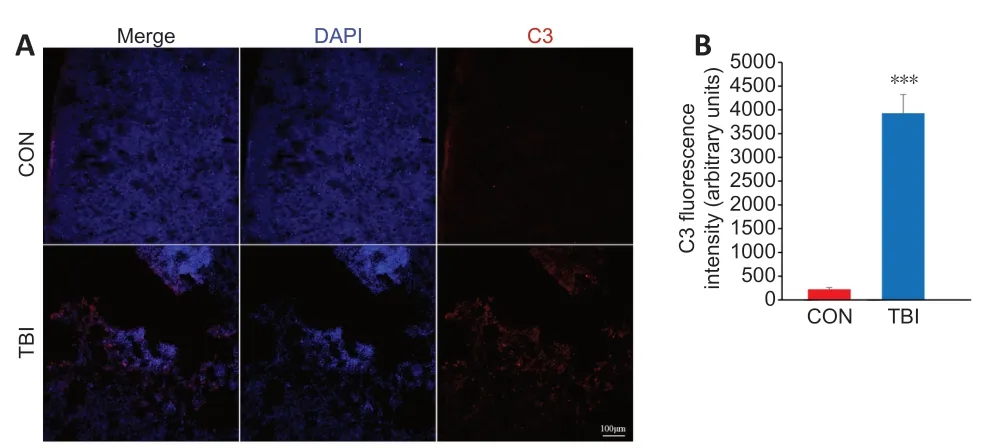
Figure 6 | Validation of the differential expression of the identified hub protein C3 in the cortex between TBI and control mice by immunofluorescence staining.
Discussion
Despite the progress that has been made in the management of TBI,its physiopathology is not completely understood,especially at the protein level (Sempere et al.,2019).To provide new insights into the nature and progression of TBI,in this study we performed global proteomic profiling in the well-established murine CCI model of TBI using TMT-based quantitative proteomics.The results showed that TBI evoked dramatic changes in the protein expression profile that were predominantly associated with inflammatory responses,including complement and coagulation cascades.Furthermore,we identified several inflammation-related TFs that may account for TBI-induced dysfunctional biologic processes.Although proteomic alteration following CCI was previously investigated,the present study differed from published studies in terms of the model species (Pumiglia et al.,2021),biosamples (Thelin et al.,2018),time points (Zhou et al.,2020),and approaches (Kobeissy et al.,2016).Use of the TMT-labeling approach has increased progressively because of its high throughput and excellent precision(Rauniyar and Yates,2014;Zhang et al.,2014).TMT-based proteomics has been used to analyze protein profiles following blast-induced or weight dropinduced TBI (Song et al.,2019;Wang et al.,2021b).In addition,TMT-labeling proteomics has been used in a rat CCI model of TBI (Zhou et al.,2020).Our study adds to this body of research by focusing on TMT-based analysis of secondary injury in a mouse model of subacute CCI.
The rapid development of omics technologies,such as transcriptomics and proteomics,has enabled high-throughput analysis of TBI-induced molecular biological alterations to be performed (Abu Hamdeh et al.,2021).Transcriptomic studies of TBI have revealed distinct changes in gene expression that are associated with multiple biological processes and provide potential targets for treatment (Samal et al.,2015;Lipponen et al.,2016;Chakraborty et al.,2021).Nevertheless,transcriptomics is limited in that it provides information about the mechanisms of diseases at the DNA and RNA levels,but not specifically at the protein level (Denny and Kalesh,2020).As proteins serve as the primary functional effector molecules in cells,proteomics can offer a more comprehensive understanding of molecular and cellular events in disease development.Several proteins involved in underlying pathophysiological processes have been identified using the proteomics technique following experimental or clinical TBI (Xu et al.,2016;Liang et al.,2019;Wang et al.,2021b;Zhang et al.,2021).However,these studies mainly focused on acute proteome alterations following TBI.Our study aimed to reveal the cortex proteomic profile in mice in the subacute stage of TBI.Because TBI is a progressive disease,and its pathophysiology varies in different stages,understanding the multitemporal pathophysiological cascade could expand the therapeutic window for TBI and help provide stage-specific therapy.
The physiopathology of TBI comprises irreversible primary injuries and secondary injuries,both of which serve as therapeutic targets (Thapa et al.,2021).Hence,understanding secondary brain injuries has become a principal concern in TBI.Neuroinflammation is considered a significant and manageable aspect of secondary injuries following TBI (Corrigan et al.,2016).TBI-induced inflammation may have detrimental or beneficial effects (Simon et al.,2017).On one hand,neuroinflammation contributes to clearances of necrotic debris,neural regeneration,repair of neuronal circuits,and reconstruction of the central nervous system barrier (Jassam et al.,2017;Clark et al.,2019;Bodnar et al.,2021).On the other hand,neuroinflammation plays a significant role in neuronal death,neuronal circuit destruction,central nervous system barriers disruption,post-traumatic epilepsy,and progressive neurodegeneration(Chen et al.,2018a;Shi et al.,2019;Bodnar,2021;Pham et al.,2021).In addition,following TBI-induced neuroinflammation,inflammatory mediators cause systemic inflammatory responses in other organs,such as the lungs,kidneys,and intestines (Sabet et al.,2021).In addition,inflammation can alter coagulation following TBI (Maegele et al.,2020).The goal of immunomodulatory therapies for TBI is to constrain the acute inflammatory response and discontinue chronic progressive neuroinflammation (Simon et al.,2017).There is a great need to identify the underlying mechanisms of TBIinduced inflammation,as well as corresponding biomarkers and therapeutic targets.
The complement system is pivotal for multiple biological progress and plays a crucial role in the neuroinflammatory response (Dinet et al.,2019;Ziabska et al.,2021).Following TBI,dysfunctional complement cascades cause secondary damage,epileptogenesis,synaptic degeneration,and cognitive deficit (Chen et al.,2020b;Alawieh et al.,2021;Mallah et al.,2021).Moreover,the complement system interferes with the coagulation cascade and homeostasis,which are closely related to TBI patient outcomes (Ricklin et al.,2010;Fletcher-Sandersjöö et al.,2020).However,the complement system also promotes neuroprotection and regeneration after TBI (Hammad et al.,2018),which suggests that the complement system is a double-edged sword in TBI pathophysiology (Ziabska et al.,2021).A better understanding of the complement system could open up new avenues for stage-specific management of TBI.
As the pivotal regulators of gene expression,TFs play crucial roles in complex biological processes (Lee and Young,2013;Zhang et al.,2020).TBI could disrupt transcription factor activity,which in turn may exacerbate brain injury by mediating secondary injuries,including inflammation (Kumar et al.,2020).In this study we found that pro-inflammatory TFs,including NFκB1,RelA,STAT1,IRF1,and Spi1,exert a notable influence on secondary injury after TBI.NF-κB1 and RelA are two of the five members of the NF-κB transcription factor family,which is a critical mediator of various biological processes such as inflammation and cell survival (Liu et al.,2017;Kumar et al.,2020).The upregulation of cortical NF-κB expression was detected in experimental and clinical TBI (Yang et al.,1995;Hang et al.,2006).Lian et al.(2012) revealed that upregulated NF-κB in astrocytes following TBI was associated with impaired inflammatory responses and worsened damage,including blood-brain barrier dysfunction and increased lesion volumes.Furthermore,Mettang et al.(2018) showed that NF-κB inhibition in neurons was associated with poorer outcomes in TBI mice.These studies indicate that NF-κB activation in neurons following TBI is beneficial,and NF-κB activation in glial cells is detrimental.In addition,activation of the HMGB1/NF-κB signaling pathway can block TBI-induced neuroinflammation (Chen et al.,2018b).Taken together,these findings suggest that NF-κB could be a potential target for TBI treatment.In addition,our earlier transcriptomic study revealed that the STAT and IRF families of TFs play critical roles in the cellular and molecular events that occur following TBI,which is consistent with the findings from the current proteomics study (Yang et al.,2021).Developing precise stage-specific therapy for TBI will require a better knowledge of the relevant TFs (Kumar et al.,2020).
There were some limitations to our study.Firstly,the sample size was small,because of both financial and ethical considerations.Secondly,given that this was a cross-sectional study that assessed one specific point in time (7 days post-TBI),we were unable to explore the possibility of a temporal relationship between TBI and neuroinflammation or complement.Additional studies involving more time points are needed to clarify the role of neuroinflammation and complement in TBI.Finally,the TMT proteomics analysis carried out as part of this study was limited to the ipsilateral cortex.Hence,future studies should validate these alterations and investigate potential changes in different brain regions.We used male mice,in accordance with a previous proteomic study (Nebie et al.,2021).Female laboratory rodents have an estrous cycle that lasts for four or five days,which is regarded as a quicker version of the human reproductive cycle (Becker et al.,2017).If female mice are at different stages in their estrous cycle,their responses during behavioral assessments will vary significantly (Estrela et al.,2021).Gender bias has long been present in animal studies,and studies that include both male and female animals are advocated (Beery and Zucker,2011).Thus,in future studies we will consider sex as a critical variable.
In summary,TBI is a biologically complex progressive disease,and its pathophysiology is far from clear.Proteomics is an ideal tool for illustrating the mechanism of TBI at the molecular and cellular levels.In this study,we used TMT-labeled proteomics and bioinformatics to identify the hub genes,pathways,and TFs in the subacute phase of TBI.The results indicated that inflammation-related biological processes,including the complement cascade,may increase anxiety after TBI.Having a comprehensive understanding of the neuroinflammatory responses that occur in TBI would advance the management of this condition.
Acknowledgments:
We would like to thank Dr.Zhong-Fang Shi from Beijing Neurosurgical Institute for her generous technical assistance.
Author contributions:
Study design:XJX,BYL and JQD;experiment studies:JQD,SHL and MSY;data acquisition:QQG and YZ;data analysis:JQD,BZ and FN;manuscript preparation:JQD;manuscript review:XJX and BYL.All authors read and approved the final manuscript.
Conflicts of interest:
The authors declare that there are no conflicts of interest associated with this manuscript.
Availability of data and materials:
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open access statement:
This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:
Renata Ciccarelli,University of Chieti-Pescara,Italy.
Additional files:
The identified proteins in 7 days post-TBI cortex tissues using a large-scale proteomics investigation.
Open peer review report 1.

- 中国神经再生研究(英文版)的其它文章
- Neuroaxonal and cellular damage/protection by prostanoid receptor ligands,fatty acid derivatives and associated enzyme inhibitors
- Extracellular vesicles in Alzheimer’s disease:from pathology to therapeutic approaches
- Molecular approaches for spinal cord injury treatment
- Sex-biased autophagy as a potential mechanism mediating sex differences in ischemic stroke outcome
- Adipose tissue,systematic inflammation,and neurodegenerative diseases
- Interleukin-1:an important target for perinatal neuroprotection?