
Hebbian plasticity:the elusive missing link at the heart of Alzheimer’s disease pathogenesis?
2022-05-25 09:27AlexanderJeans
中国神经再生研究(英文版) 2023年1期
Alexander F.Jeans
The amyloid cascade hypothesis of Alzheimer’s pathogenesis:
The amyloid cascade hypothesis of Alzheimer’s disease (AD) pathogenesis will shortly celebrate its thirtieth birthday (Hardy and Higgins,1992).Based on abundant genetic and biochemical evidence,it proposes that deposition of the amyloid-β (Aβ) peptide in brain parenchyma is an essential upstream trigger in AD pathogenesis that drives a cascade of events,specifically including the recruitment and pathological hyper-phosphorylation of the micro-tubule-associated protein tau,that culminate in derangement of synaptic function and,eventually,neuronal death.Although the hypothesis has been challenged many times over the last three decades,principally based on a number of observations of AD pathology and clinical progression that it appears not to readily explain (Makin,2018),its fundamental assertions that Aβ deposition is a critical early event,and that this somehow leads to the later recruitment of hyperphosphorylated tau,still appear to hold true (Selkoe and Hardy,2016).Therefore,and in spite of its difficulties,the amyloid cascade hypothesis,albeit slightly refined and qualified over the years,is still the dominant model of AD pathogenesis.The central importance of tau to the disease process has been confirmed by a number of more recent studies that demonstrate convincingly that tau is essential for many of the canonical ADassociated synaptic and behavioral phenotypes,which can be rescued in animal models of AD by tau knockout (Mucke and Selkoe,2012).However,despite its clear significance in AD pathogenesis,the cellular mechanism by which Aβ recruits tau to bring about synaptic and cognitive decline has remained obscure.Dysfunctional Hebbian plasticity links Aβ to tau
in a model of AD:
One of the most important and best-studied AD-related phenotypes associated with exposure to pathogenic forms of Aβ,in particular the oligomeric forms of Aβ (Aβ) that are particularly potent in pathogenesis,is the modulation of Hebbian synaptic plasticity processes.These changes have been best studied at the CA3-CA1 (Schaffer collateral)synapse in the hippocampus,where exposure to Aβcauses a loss of synaptic long-term potentiation and facilitation or enhancement of long-term depression(LTD).We were initially intrigued by the finding that these effects are mediated via an extra-synaptic population of postsynaptic N-methyl-D-aspartic acid(NMDA)-type glutamate receptors (Mucke and Selkoe,2012),because extra-synaptic NMDA receptors are particularly efficiently activated at synapses with a high probability of release (Pr) (Lozovaya et al.,1999),an index of how efficiently action potentials are converted to neurotransmitter release events at the presynaptic terminal.Several studies have reported increases in Pr following exposure to Aβ(Taylor et al.,2021) and we,therefore,formulated a hypothesis that in the context of AD,enhancements in neurotransmitter release probability might drives greater activation of extra-synaptic glutamate receptors and consequently changes in Hebbian plasticity,specifically the enhancement of LTD.A pointer towards a potential link between altered plasticity and tau came with a more recent study that demonstrated that under physiological conditions,the induction of LTD recruits and phosphorylates tau,albeit at just two sites (Regan et al.,2015).Accordingly,we extended our hypothesis to suggest that the enhanced LTD seen following Aβexposure might similarly recruit and phosphorylate tau,but perhaps to an even greater degree than seen during normal LTD.To test our hypothesis,we began by examining the action potential-evoked release of previously loaded fluorescent FM dyes from presynaptic terminals of hippocampal CA3-CA1 synapses in rodent organotypic hippocampal slices.These experiments showed faster dye unloading following treatment with Aβ,confirming increased Pr (Taylor et al.,2021),and we then turned to acute hippocampal slices to show that Aβalso drive an enhancement in the magnitude of CA3-CA1 LTD induced by low-frequency synaptic stimulation,which has been previously reported.However,we also showed that normalization of probability of release by partial inhibition of presynaptic voltage-gated Cachannels can rescue the enhanced LTD,restoring it to a normal level.We then wished to study the link between Aβ-enhanced LTD and tau phosphorylation.As these next experiments involved more chronic Aβexposure,we returned to using organotypic slices to firstly show that tau hyperphosphorylation,which we assessed using an antibody recognizing phosphorylation at specific pathologically-relevant tau residues,was enhanced following Aβtreatment,and that this also could be rescued by either partial blockade of presynaptic Ca channels to normalize Pr,or by blockade of the NMDA receptors that are responsible for LTD induction.While together,these results suggest that Aβ-mediated increases in Pr tend synapses towards inappropriate or excessive induction of LTD,which in turn could promote supraphysiological phosphorylation of tau,they do not amount to a direct demonstration of causation.To confirm a causal relationship between enhanced LTD induction and hyper-phosphorylation of tau,we imposed extreme LTD-inducing conditions on organotypic slices and asked whether this alone,in the absence of Aβ,would be sufficient to drive the hyper-phosphorylation of tau.Indeed we found that this was the case,both with chemical LTD induction protocols and with protracted optogenetic lowfrequency synaptic stimulation.Our results,therefore,support a possible causal chain in which Aβincreases neurotransmitter release probability,thus leading to enhanced LTD induction,which in turn drives hyper-phosphorylation of tau(Figure 1
).While this study identifies a potential mechanistic pathway linking the two critical pathogenic proteins of AD,it has limitations that will need to be addressed in future work,an important one being that our work was carried out entirelyin vitro
,and it will be important to confirm the findings inin vivo
systems.Importantly,this would circumvent the potential issue of having used one particular preparation of Aβ,which may not represent all of the pathogenic species present in the intact brain.There is also molecular mechanistic detail missing,notably the identity of the specific kinases that mediate the tau phosphorylation events downstream of LTD induction.Identifying these key players in the process raises the possibility of new candidate therapeutic targets,a very welcome possibility in AD,where the lack of such targets currently constitutes a major roadblock to therapeutic progress.Finally,it is important to note that,although this mechanism is likely to be important in the recruitment of tau to the disease process,once hyperphosphorylated pathological tau begins to accumulate it may no longer be dependent on LTD or even on Aβat all to progress,as there is now good evidence of multiple Aβ-independent mechanisms regulating tau pathology in AD (van der Kant et al.,2020).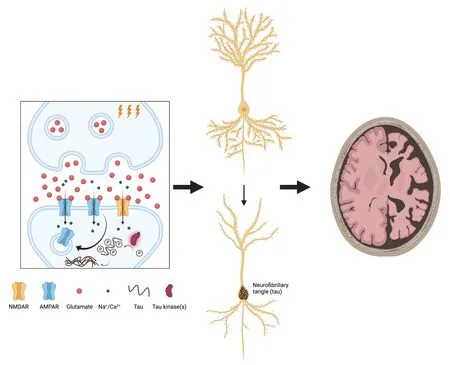
Figure 1 | Hebbian plasticity links Aβ to tau in a model of AD.
Understanding tau in AD:an experimental challenge with a potential solution:
One of the most significant strengths of our study is that it examines only native,endogenous tau within our slice culture model,and the study design deliberately avoids the use of exogenous tau.This is,fortunately,sometimes possible in studies such as ours,which focus on events in the pathological cascade upstream of tau hyper-phosphorylation or deposition,and may be able to use the phosphorylation or aggregation status of endogenous tau as a readout.However,this is very often not feasible when studying tau in the disease context,and investigators will have no alternative but to introduce some form of pathological tau to their experimental system to generate a model suitable for their work.Such introductions of exogenous tau have enabled numerous valuable studies in the tauopathy field,and the approach can have great merit.However,it can be troublesome when employed in the study of AD,which is not a primary tauopathy,as it incorporates a number of critical assumptions about the nature of AD-associated pathological tau that are frequently not acknowledged but which,if not properly considered,can critically confound experimental outcomes.To understand why this is,it is necessary first to understand the nature of the models that are generally used to investigate neurodegenerative disease.Most of these models,and particularly the animal models,are derived from knowledge of the genetic basis of these conditions in human populations;specifically,from the identification of rare familial,early-onset but otherwise clinicopathologically typical variants of diseases that are more usually sporadic in their occurrence.These inherited forms usually result from a single mutated gene that can readily be expressed as a transgene in a model biological system,in contrast to the common,sporadic forms of these diseases which usually have only a relatively limited genetic basis as identified by genome-wide association studies.Examples of this approach to disease modeling include expressing α-synuclein transgenes carrying a pathogenic mutation to generate mouse models of Parkinson’s disease,and mutated amyloid precursor protein (APP) and presenilin-1 (PS1) transgenes to model AD in mice.However,there is an important caveat with regard to these genetic models of AD,as they do not develop the hyperphosphorylated tau pathology that is characteristic of the disease (Elder et al.,2010),which makes the study of disease-associated tau very difficult.The reasons for this lack of tau pathology may be many,one of the most likely being the limited lifespan of model organisms,which may not allow extracellular tau tangles time to develop.In any case,it means that many of the commonly used AD model mice do not recapitulate one of the central elements of the disease phenotype.In order to remedy the absence of tau deposits in mice carrying APP and/or PS1 transgenes,a variety of approaches have been used based on the introduction of exogenous tau by either genetic or physical (direct inoculation) means.The former approach,which has been by far the most commonly used,is particularly problematic.It typically involves the expression of disease-relevant mutant APP and/or PS1 alongside a tau transgene bearing one of a number of pathogenic mutations associated with tau deposition and neurodegeneration (Elder et al.,2010).A variety of such pathogenic tau mutations have been described in several families.However,none of these has ever been associated with the development of AD.Instead,they cause clinicopathological syndromes that are quite distinct from AD:frontotemporal dementia with Parkinsonism linked to chromosome 17 or,much less commonly,progressive supranuclear palsy or Pick’s disease (Wang and Mandelkow,2016).In these diseases,the pathological tau tissue deposits almost always contain just one of the two major tau splice isoform classes,which are three and fourrepeat tau (Wang and Mandelkow,2016).Critically,however,tau in AD does not have this composition but is mixed three and four-repeat,suggesting that it is biochemically distinct from the tau deposits of primary genetic tauopathies,and therefore very likely to be recruited via a different pathway.Nonetheless,these mouse models have been used in a multitude of AD studies over many years;for example,one of the earlier models (Oddo et al.,2003) now has over 4000 citations.Data from these animals,particularly with regard to the role of tau in AD,therefore must be interpreted with extreme caution because of this key confounding issue.
Within the last few years,new structural data has emerged that both substantially endorse the above concerns around using potentially inappropriate genetic models of tauopathy to understand tau in AD,yet also suggests a way forward to begin to resolve this problem.A series of papers employing the powerful technique of cryo-electron microscopy has resolved the structures of protofibrillar tau tissue deposits in a variety of tauopathies including AD.The authors find clear differences in structures within protofibrils in each disease,suggesting that there may be a link between the molecular conformation of tau deposits and the resulting clinicopathological phenotype.These structures have been used to formulate a novel classification of the tauopathies(Shi et al.,2021),which confirms that AD is a distinct entity from the diseases caused by mutations in tau.However,it also suggests that we may have the means to create faithful,fully disease-specific models of tauopathies if we use a direct inoculation approach.Such an approach has been shown to be successful in creating valid disease models that recapitulate both the histology and progression of human AD tau pathology.Initial attempts used tau derived from tau transgenic mice,which was able to seed the formation of aggregates when injected into the brains of mice over-expressing a non-mutated human tau transgene.Not only this,these aggregates were able to spread to anatomically connected brain regions.This finding was later extended to show that the same outcomes were possible using samples of tau prepared from human tauopathy patient brains,and that these could even propagate in non-transgenic mice.Furthermore,the inclusions formed following these injections had the characteristic morphology of the relevant tauopathy,for example,injections of tau from argyrophilic grain disease brains yielded argyrophilic grains and neurofibrillary tangles,while injections of tau from corticobasal degeneration brains resulted in neuropil threads and a small number of silverstaining inclusions in neuronal bodies,as is observed in the human disease (Clavaguera et al.,2013).While these studies concerned only primary tauopathies,the same formation of hallmark inclusions with spread of pathology was later accomplished using AD brain-derived tau in non-transgenic mice (Guo et al.,2016).This of course raises the question of how interregional spread of AD tau pathology on a wild-type background might be reconciled with the Aβ-initiated,Hebbian plasticity-driven hyper-phosphorylation of tau that we report.There are several possibilities,one of which is that the spread of tau pathology is somehow associated with increased Aβ production,another being that the plasticity-driven mechanism is critical in early disease stages,but once sufficient hyperphosphorylated tau has been formed it can then propagate and spread autonomously.Such a mechanism,in which Aβ is only critical for the initiation,but not the later spread,of AD pathology has in fact been proposed to account for the apparent failure of anti-amyloid therapies in clinical trials,where they have only been given once the disease is established.
Together,all of these findings raise the possibility that the direct inoculation approach could be used to develop a series of highly specific and pathophysiologically relevant mouse models of all of the tauopathies,including sporadic diseases such as AD.In principle,a single inoculation of the structural form of tau associated with a particular disease could initiate tau pathology which would then spread through the brain,although whether this would follow the same patterns as the corresponding human diseases indefinitely remains to be seen.It is also true that a substantial amount of further work on the characterization and optimization of such models would be needed before they could be made ready for routine research use.Nonetheless,the approach holds the prospect not only of unparalleled pathological fidelity to human disease but also of a dynamic disease model which may potentially,unlike transgenic animals expressing a mutated gene in dispersed cell populations or even ubiquitously,be able to recapitulate the temporal and anatomical progression of tau pathology from an initial focus to surrounding areas in a mostly predictable way,as described in human brains by the Braak staging system (Wang and Mandelkow,2016).
Alexander F.Jeans
Department of Pharmacology,University of Oxford,Oxford,UK
Alexander F.Jeans,MB,DPhil,alexander.jeans@pharm.ox.ac.uk.https://orcid.org/0000-0002-4004-3676(Alexander F.Jeans)
Date of submission:
October 29,2021Date of decision:
December 30,2021Date of acceptance:
January 12,2022Date of web publication:
May 31,2022https://doi.org/10.4103/1673-5374.340402
Jeans AF (2023) Hebbian plasticity:the elusive missing link at the heart of Alzheimer’s disease pathogenesis? Neural Regen Res 18(1):123-124.
Availability of data and materials:
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open access statement:
This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewers:
Abraam M.Yakoub,Stanford University,USA;Hans-Gert Bernstein,Otto-von-Guericke University Magdeburg,Germany.
Additional file:
Open peer review report 1.

- 中国神经再生研究(英文版)的其它文章
- Neuroaxonal and cellular damage/protection by prostanoid receptor ligands,fatty acid derivatives and associated enzyme inhibitors
- Extracellular vesicles in Alzheimer’s disease:from pathology to therapeutic approaches
- Molecular approaches for spinal cord injury treatment
- Sex-biased autophagy as a potential mechanism mediating sex differences in ischemic stroke outcome
- Adipose tissue,systematic inflammation,and neurodegenerative diseases
- Interleukin-1:an important target for perinatal neuroprotection?