
Initial failures of anti-tau antibodies in Alzheimer’s disease are reminiscent of the amyloid-β story
2022-05-25 09:26BrunoImbimboClaudiaBalducciStefaniaIppatiMarkWatling
中国神经再生研究(英文版) 2023年1期
Bruno P.Imbimbo,Claudia Balducci,Stefania Ippati,Mark Watling
Tau is an important protein of the central nervous system formed by 352–441 amino acids and encoded by theMAPT
(microtubule-associated protein tau)gene on chromosome 17 which generates 6 isoforms.Tau is located in axons,dendrites,nucleus,cell membrane,and synapses of neurons.The protein is also expressed to a lesser extent in astrocytes and oligodendrocytes,although its role in these cells has been little investigated.The protein is also present in the interstitial fluid and can cross into the cerebrospinal fluid (CSF) and reach the systemic circulation.The main function of tau is promoting the assembly and stabilization of microtubules in neuronal axons.Tau plays also a role in a range of other biological processes including myelination,neurogenesis,motor function,learning,and memory (Kent et al.,2020).The binding of tau to microtubules is regulated by its phosphorylation/dephosphorylation equilibrium.In physiological conditions,tau is unfolded and phosphorylated,while the pathological form is characterized by an excess of hyperphosphorylation leading to disengagement from the microtubules,and conformational changes that lead to the formation of paired helical and straight filaments of abnormally phosphorylated tau and subsequently to tau aggregates.These aggregates can cause degeneration of neurons and glial cells that ultimately lead to various clinical cognitive,behavioral,and motor manifestations,which are classified into different types of neurodegenerative disorders called‘tauopathies’.Tauopathies are classified into primary and secondary tauopathies.In primary tauopathies,the abnormal tau accounts for the primary underlying neurodegenerative process.Primary tauopathies include progressive supranuclear palsy (PSP),corticobasal degeneration,corticobasal syndrome tauopathy,Pick’s disease,frontotemporal dementia,frontotemporal lobar degeneration,primary progressive aphasia,MAPT
mutation,argyrophilic grain disease,and primary age-related tauopathy.In secondary tauopathies,tau neuronal inclusions occur in association with the extracellular deposition of a second aggregated protein.Secondary tauopathies include Alzheimer’s disease (AD) and Down syndrome(in which amyloid-beta [Aβ] accumulates),Lewy body dementia (in which α-synuclein accumulates),and chronic traumatic encephalopathy (in which TAR DNAbinding protein 43) accumulates.Recently,it has been shown that tau is differentially phosphorylated in various tauopathies.In the brain of AD patients,there is increased phosphorylation at positions Ser202,Thr231,and Ser235,while Pick’s disease brains show increased phospho-Ser202,and argyrophilic grain dementia brains show increased phospho-Ser396.In neurodegenerative tauopathies,pathological tau can propagate between neuroanatomically connected brain regions by multiple mechanisms,spreading tau pathology throughout the brain.However,recent neuropathological studies in the AD brain suggest that local replication,rather than spreading between brain regions,is the main process driving the overall rate of tau accumulation in neocortical regions.There are also contrasting theories as to whether it is soluble or aggregated tau species that correlate with disease progression and cognitive decline in AD patients.
No drugs have yet been approved for the treatment of primary tauopathies.The treatments currently used are mostly off-label medications targeting symptomatic management,with only minimal evidence from a few small,controlled trials to support their use.However,recent advances in the clinical,neuropathological,and biochemical characterization of tauopathies have prompted the search for disease-modifying therapies.In the last 15 years,the search for drugs interfering with pathological aggregation,processing,and accumulation of tau has been particularly intense. More than 30 drugs have reached the clinic including two tau aggregation inhibitors,three microtubule stabilizers,three glycogen synthase kinase-3β inhibitors,one tau acetylation inhibitor,three O-GlcNAcase inhibitors,two anti-tau active vaccines,11 anti-tau monoclonal antibodies,one tau antisense oligonucleotide,and one progranulin enhancer (Figure 1A
).Unfortunately,to date none of these pharmacological approaches have produced clinical benefits in patients.Major efforts have been devoted to the development of passive immunotherapeutic approaches to stimulate brain clearance of tau and phosphorylated tau aggregates,and several anti-tau monoclonal antibodies directed against different tau epitopes (Figure 1B
) have also entered trials.Studies in animals have suggested that the therapeutic efficacy of tau immunotherapy may depend on the precise tau region that is targeted,with behavioral effects not directly correlating with the reduction in tau pathology burden.Up to now,controlled trials of anti-tau antibodies in both primary(mainly progressive supranuclear palsy) and secondary(mainly AD) tauopathies have failed to show clinical efficacy.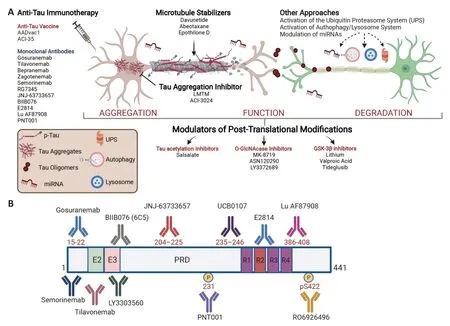
Figure 1 | Scheme showing point of attack of different pharmacological classes of anti-tau drugs and epitope locations on the primary sequence of tau,for antibodies currently in clinical trials.
Among 12 anti-tau therapies in development for AD,a Phase 2 trial of semorinemab,a humanized antitau antibody against the N-terminal epitope,showed no differences in the rates of cognitive decline of prodromal or mild AD patients compared with placebo(Mullard,2021).Gosuranemab,another monoclonal antibody targeting the N-terminal region of tau,even worsened cognitive decline in patients with early stages of AD (Shulman et al.,2021).The development of zagotenemab,whose primary epitope is in tau’s N-terminal region,was recently discontinued due to its failure to positively affect the primary endpoint in a Phase 2 study in early AD.Thus,all monoclonal antibodies targeting the N-terminal region of tau (semorinemab,gosuranemab,tilavonemab,zagotenemab) have failed in major clinical trials in AD or other tauopathies.The reasons for these failures are not clear,but we know that the N-terminal domain of tau provides spacing between the microtubules and this activity may be vital for the physiological function of tau.Thus,attacking this specific epitope of tau with selective antibodies may not produce clinical benefits.The scientific community is now directing its efforts to other regions of the tau protein.Midregion antibodies are believed to have more chance of interfering with the cell-to-cell propagation of pathogenic,aggregated tau than do N-terminal anti-tau antibodies.More recently it has been proposed that antibodies binding the microtubule-binding region,which spans residues 224–369,may be better at preventing aggregates from spreading (Horie et al.,2021) and several such antibodies have now entered Phase 1 or 2 clinical testing.
Failed clinical trials with anti-tau monoclonal antibodies in AD have provided potentially important information for future investigations.In a failed trial of semorinemab in 457 subjects with early AD (TAURIEL study),the antibody elicited maximal increases in plasma tau,indirectly indicating a possible transfer of soluble tau from CNS to plasma,but did not slow tangle accumulation in brain as measured with the tau PET tracer guanosine triphosphate 1,relative to placebo.In another failed trial of gosuranemab in 654 subjects with early AD (TANGO study),the drug was shown to engage its target because the CSF level of N-terminal tau fragments fell in the active treatment groups but,again,no effect on tau accumulation was detected by tau-PET scans and accelerated cognitive decline was seen to occur (Shulman et al.,2021).Interestingly,another trial of semorinemab in 273 subjects with moderate AD (LAURIET study) produced a signal of partial efficacy with a 42% reduction in the rate of cognitive decline as measured by the Alzheimer’s Disease Assessment Scale,Cognitive Subscale,11-item Version from baseline to 49 weeks compared to placebo.On the other hand,the antibody did not show any improvement relative to placebo on the other co-primary functional endpoint,the Alzheimer’s Disease Cooperative Study-Activities of Daily Living.It also did not improve secondary endpoints assessing cognition with the Mini-Mental State Examination,or global performance as measured by the Clinical Dementia Rating-Sum of Boxes (Monteiro et al.,2021).Unfortunately,it is not made clear whether the drug affected tau-PET.Nevertheless,these findings do suggest that antitau immunotherapies that reduce soluble tau species in the brain during the early phases of AD do not produce significant clinical benefits.
Anti-tau monoclonal antibodies (tilavonemab and gosuranemab) have also failed to show clinical efficacy in placebo-controlled clinical trials in PSP,a prototypical primary neuropathy in which tau is believed to play a causative role.A 1-year,Phase 2 trial of tilavonemab failed to show beneficial effects in a study involving 378 subjects with PSP(Hӧglinger et al.,2021).The study with tilavonemab was terminated earlier than anticipated at an interim evaluation by virtue of futility in changing the primary efficacy endpoint,the Progressive Supranuclear Palsy Rating Scale (PSPRS).PSPRS scores trended toward worsening in patients on the higher dose oftilavonemab (4000 mg) compared to the placebo arm.Previous work in transgenic mice expressing human tau had shown that this antibody slowed progression of tau pathology,reduced loss of brain volume,and augmented cognitive performance.In the Phase 2 study,target engagement by tilavonemab was shown through lower free tau levels in CSF and higher levels in plasma,compared with placebo.Nevertheless,the trial was stopped early due to prespecified futility criteria,indicating that efficacy was unlikely to be demonstrated.In the 1-year gosuranemab trial involving 486 participants,a positive effect of the monoclonal antibody on PSPRS was not shown in spite of the unbound N-terminal tau levels in CSF decreasing by 98% with gosuranemab and increasing by 11% with placebo (Dam et al.,2021).
If these negative results are confirmed with other anti-tau antibodies or antisense oligonucleotides,we have to conclude that lowering soluble tau does not produce clinical benefits in either PSP or AD.This may mean that the tau contribution to neurodegeneration is due to a loss of its function at the microtubules.This would mean that soluble tau is not causing clinical symptoms and represents an epiphenomenon of the true pathophysiological process.However,some investigators have suggested that these clinical failures can be overcome through better trial designs(factorial design for testing combinations of antitau and anti-Aβ drugs),better clinical measures(more sensitive composite scales),better tau epitopes (central),or better tau species (pathogenic conformers) (Shugart et al.,2021).This situation is reminiscent of that which we have experienced over 20 years of research with anti-Aβ drugs.The initial clinical failures were ascribed to the lack of drug selectivity of drugs for“toxic”oligomeric Aβ species or to the advanced stage of the disease (mild-tomoderate instead of early or pre-clinical stages),to the enrollment of patients without biomarker proven Aβ pathology,to the insensitivity of cognitive and clinical scales (recommending the introduction of composite scales) or to testing the drugs in sporadic AD rather than in autosomal dominant AD.This long history of methodological and technical adjustments to clinical trials in AD has,however,continued to yield poor results.The best results of these experimental refinements have recently been obtained with the anti-Aβ monoclonal donanemab.In an 18-month Phase 2 study involving 257 patients with early AD donanemab was shown to improve by only 3 points,compared to placebo,a composite scale (iADRS or integrated Alzheimer’s Disease Rating Scales) that has a range of 0 to 144 points and a baseline score of 106 points (Mintum et al.,2021) -this is clearly a trivial clinical benefit.
A Phase 2 study with lecanemab,where an adaptive design was adopted,apparently produced encouraging results.The study was conducted with an adaptive design but a change of the clinical protocol to exclude carriers of the epsilon4 allele of the apolipoprotein E gene (APOE4
) carriers at the highest doses generated an imbalance in the number ofAPOE4
carriers with only 30% of the treatment cohort beingAPOE4
carriers compared with 71% of the placebo group.SinceAPOE4
carriers generally experience faster cognitive decline,the placebo group would be expected to decline more quickly than the lecanemab group,thus ‘favoring’ the latter.Despite this potential advantage for lecanemab,the study still did not meet the 12-month primary endpoint.Nevertheless,the authors claimed a consistent reduction in clinical decline across several clinical and biomarker endpoints that is difficult to interpret in view of the above (Swanson et al.,2021).Nevertheless,in June 2021,the Food and Drug Administration designated lecanemab a breakthrough therapy,thus expediting its regulatory review and in December 2021,the agency also granted“fast track”designation for this monoclonal antibody.We hope that the same journey made with anti-Aβ drugs will not be repeated with anti-tau drugs.In the case of tauopathies,consideration must be given to shifting research efforts from a gain-of-function model where tau is targeted by enhancing its clearance,to a loss-of-function model where normal tau levels and function are restored.We know that in AD and in other tauopathies,protein transition into insoluble aggregates and deposits implies a corresponding decrease in the pool of soluble species that is associated with a loss of their normal physiological functions (Kent et al.,2020).Thus,removing soluble Aβ and tau may only exacerbate clinical symptoms,as has been observed with β-secretase and γ-secretase inhibitors in AD.The same can happen with anti-tau antibodies that can deplete physiological levels of soluble tau in the brain.Some anti-Aβ monoclonal antibodies may have marginal beneficial effects in early stages of AD possibly due to their ability to mobilize soluble Aβ from brain amyloid plaques.The small positive effects observed with donanemab in early AD (Mintum et al.,2021) may be due to the fact that the study protocol stipulated that during treatment with donanemab the dose of the drug should be reduced if the amyloid plaques were markedly reduced (between 25 and 11 centiloids)or even interrupted in case of massive reduction in amyloid plaques (less than 11 centiloids).This approach avoided excess depletion of physiological levels of soluble Aβ in the brain due to prolonged and complete removal of amyloid plaques.
In conclusion,it is our opinion that we should question the assumption that in AD or other tauopathies Aβ deposits or tau aggregates represent causative biomarkers of disease,and that the elimination of these would stop or slow disease progression.Previous studies have shown that there is a lack of correlation between clinical symptoms or neuronal loss and brain Aβ burden in AD and between clinical symptoms or neuronal loss and tau in PSP.The most convincing argument against the thesis that brain pathology findings in AD or other tauopathies explain clinical symptoms arises from clinical trials of putative disease-modifying drugs that significantly reduce aggregate pathology but do not elicit clinical benefit– indeed,in some cases even causing worsening symptoms (Panza et al.,2019).While most of the pharmacological evidence for this has come from anti-Aβ approaches in AD over the past two decades,in recent years early evidence has been accumulating that anti-tau interventions similarly do not work in tauopathies.Tau accumulation is a terminal event in neurodegenerative disorders,and because the increase of CSF tau in patients with AD occurs closer to the onset of neurodegeneration than does the decrease of Aβ,tau has been shown to correlate to a greater extent with dementia than Aβ,with tau aggregates predicting dementia better than Aβ deposits (Ossenkoppele et al.,2021).Despite a huge series of negative Phase 2 and 3 clinical trials with anti-Aβ drugs in AD (Panza et al.,2019),the conviction that the accumulation of supposedly toxic proteins(Aβ and tau) in the brain of AD patients causes clinical symptoms has continued to drive massive discovery pipelines.It is time to reconsider subjecting patients to the removal of physiological levels of soluble Aβ and tau and to concentrate our efforts on understanding why,in AD and tauopathies,Aβ and tau begin to aggregate in the brain to form deposits.The authors declare that they have no conflicts of interest.No conflicts of interest exist between TranScrip Ltd.and publication of this manuscript
.Bruno P.Imbimbo,Claudia Balducci,Stefania Ippati,Mark Watling
Department of Research &Development,Chiesi Farmaceutici,Parma,Italy (Imbimbo BP)
Department of Neuroscience,Istituto di Ricerche Farmacologiche“Mario Negri”IRCCS,Milan,Italy(Balducci C)
San Raffaele Scientific Institute,San Raffaele Hospital,Milan,Italy (Ippati S)
CNS &Pain Department,TranScrip Ltd.,Reading,UK (Watling M)
Bruno P.Imbimbo,PhD,b.imbimbo@chiesi.com.http://orcid.org/0000-0002-0327-7262(Bruno P.Imbimbo)
Date of submission:
November 30,2021Date of decision:
January 11,2022Date of acceptance:
January 24,2022Date of web publication:
May 31,2022https://doi.org/10.4103/1673-5374.340409
Imbimbo BP,Balducci C,Ippati S,Watling M (2023) Initial failures of anti-tau antibodies in Alzheimer’s disease are reminiscent of the amyloid-β story.Neural Regen Res 18(1):117-118.
Open access statement:
This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Neuroaxonal and cellular damage/protection by prostanoid receptor ligands,fatty acid derivatives and associated enzyme inhibitors
- Extracellular vesicles in Alzheimer’s disease:from pathology to therapeutic approaches
- Molecular approaches for spinal cord injury treatment
- Sex-biased autophagy as a potential mechanism mediating sex differences in ischemic stroke outcome
- Adipose tissue,systematic inflammation,and neurodegenerative diseases
- Interleukin-1:an important target for perinatal neuroprotection?