
Crosslink between mutations in mitochondrial genes and brain disorders:implications for mitochondrialtargeted therapeutic interventions
2022-05-25 09:26JaspreetKalra
中国神经再生研究(英文版) 2023年1期
Jaspreet Kalra
Abstract At the present,association of mitochondrial dysfunction and progression of neurological disorders has gained significant attention.Defects in mitochondrial network dynamics,point mutations,deletions,and interaction of pathogenomic proteins with mitochondria are some of the possible underlying mechanisms involved in these neurological disorders.Mitochondrial genetics,defects in mitochondrial oxidative phosphorylation machinery,and reactive oxygen species production might share common crosstalk in the progression of these neurological disorders.It is of significant interests to explore and develop therapeutic strategies aimed at correcting mitochondrial abnormalities.This review provided insights on mitochondrial dysfunction/mutations involved in the progression of Alzheimer’s disease,Huntington’s disease,and epilepsy with a special focus on Parkinson’s disease pathology.Along with the deleterious effects of mitochondrial mutations in aforesaid neurological disorders,this paper unraveled the available therapeutic strategy,specifically aiming to improve mitochondrial dysfunction,drugs targeting mitochondrial proteins,gene therapies aimed at correcting mutant mtDNA,peptidebased approaches,and lipophilic cations.
Key Words:adenosine-triphosphate deficiency;mitochondrial fission/fusion;mitochondrial mutations;neurodegenerative disorders;oxidative phosphorylation;therapeutic interventions
Introduction
Among all organs,the brain occupies approximately 2% of the total body weight percentage.Owing to its increased weight,approximately 15% of the total cardiac output is received by the brain;furthermore,it also utilizes a major portion approximately 20% of the total oxygen produced by the body.Excessive energy requirements of the brain are crucial for the proper functioning of neurons and maintenance of ionic gradient across the plasma membrane.Impairment in oxygen demand and supply cycle leads to neuronal cell death (Zhu et al.,2018).Apart from being a major source for adenosinetriphosphate (ATP) production,mitochondria are necessary for regulating homeostasis and cell survival.However,defects in mitochondrial function are associated with the increased incidence of cell death and reactive oxygen species (ROS) generation (Bhatti et al.,2017).Nevertheless,mitochondrial ROS are implicated in inducing mutations in mitochondrial DNA (mtDNA) and play a key role in the development and progression of neurological disorders.
Search and Selection Criteria
In this narrative review,PubMed database was searched for articles published from 1988 to 2021 using keywords such as neurological disorders,neurodegenerative disorders,mitochondrial dysfunction,mutations in mitochondria,parkin,α-synuclein,PARK1,therapies targeting mitochondrial dysfunction,and antioxidant therapies.Other than PubMed,official websites such as https://doi.org/10.19723/j.issn.1671-167X.2020.05.009,https://www.ninds.nih.gov/Disorders/All-Disorders/Huntingtons-Disease-Information-Page,https://www.verywellhealth.com/the-4-as-of-alzheimers-disease-98591,were also used for data retrieval and collection.
Genetics of Mitochondria
Defects in mitochondrial oxidative phosphorylation machinery,reactive oxygen species production,and neurological disorders
Mitochondria maintain cellular homeostasis by producing numerous redox enzymes.Under stress conditions,it augments massive ROS production,influences the overall mitochondrial health and quality control function.Even though multiple cellular compartments are associated with ROS production,a majority of the intracellular ROS is delivered back to mitochondria (Rose et al.,2017).This process is well recognized as mitochondrial oxidative phosphorylation (OXPHOS).OXPHOS machinery is located on the inner surface of the mitochondrial membrane and composed of protein complexes having five multi-subunits.Each multi-subunit in respiratory chain complexes are encoded by the nucleus that is commonly rereferred as:(complex I),(complex II),(complex III),(complex IV),and ATP synthase (complex V).When compared with all of the respiratory chain complexes,complex I consists of a maximum number of mitochondrial encoded proteins (seven polypeptide chains) (Table
1
).Abnormalities in mitochondria function are the major determinants for various apoptotic pathways,dysregulation of metabolic hemostasis,and fusion/fission process.Inability to produce ATP,defects in mitochondrial function may progress towards deleterious mutations in mitochondria.As per the mitochondrial free-radical theory,mitochondria produce excessive ROS in aging (Nunnari and Suomalainen,2012).Elevated oxidative load causes an imbalance in proper mitochondrial functioning,ultimately leading to increased cell death.Excessive ROS in turn stimulate the cytosolic signaling molecules crucial for initiating intrinsic mitochondrial apoptotic cascade(Suomalainen and Battersby,2018).
Table 1 | Encoding of protein subunits in respiratory enzyme complexes:component of oxidative phosphorylation
Mitochondrial DNA
mtDNA is a circular-shaped double-stranded molecule of 16.569-kb having 37 genes.Approximately 13 structural genes encode for respiratory chain subunits while the remaining 24 genes are divided into 2 rRNA genes and 22 tRNA genes,though many of them are encoded by the nuclear genome (Kotrys and Szczesny,2019).mtDNA is highly susceptible to mutational alterations.Compared with nuclear DNA (nDNA),mtDNA is 10 times more susceptible to mutations and associated changes.Studies have confirmed the presence of antioxidant and DNA repair enzymes such as 8-oxoguanine DNA glycosylase(OGG1) and mutY DNA glycosylase in mitochondria,which exhibit preventive potential against DNA mutation.Unlike nDNA,mtDNA is not layered by histones (histones act as a sheath for protection),which makes mtDNA more vulnerable to damage (Kaarniranta et al.,2019).Balanced mitochondrial quality control is required for maintaining effective communication with a nucleus.OXPHOS causes a significant increase in ROS-mediated damage that initiates retrograde signaling in the mitochondrial pathway.Stimulation of retrograde signaling often activates an adaptive nuclear response that leads to impairment in mtDNA and calcineurin-dependent activation of NFκB.Long mitochondrial disruption has been known to be associated with the expression of more than 40 nuclear genes (Chowdhury et al.,2020).The relevance of mutations in mtDNA could be further understood by knowing the basic differences between nuclear and mitochondrial genetics.Table 2 describes the genetic classification of selected mitochondrial diseases (Kazak et al.,2012).
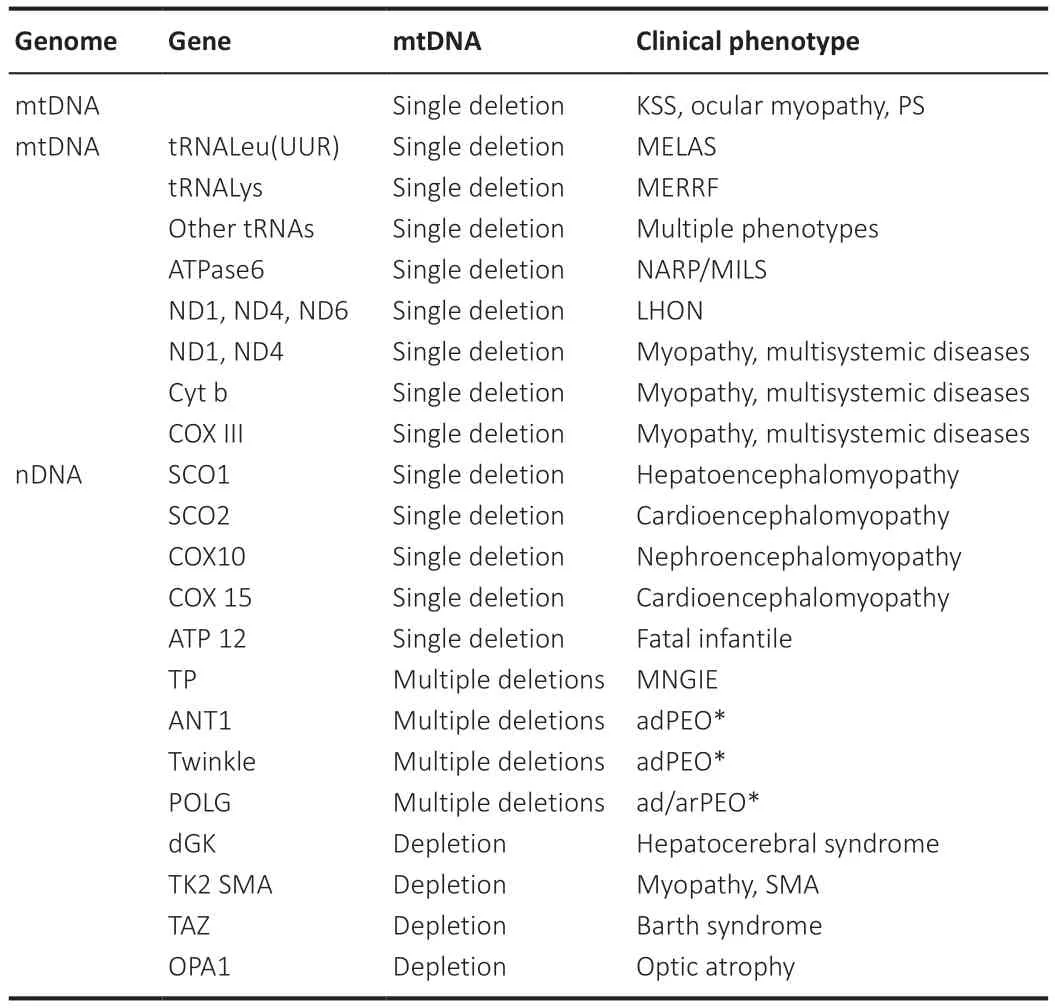
Table 2 | Classification of the more common forms of mitochondrial diseases on the basis of genetics
Crosstalk between reactive oxygen species and mitochondrial DNA dysfunction
Accumulating evidence indicates the implication of ROS in mtDNA dysfunction.Mitochondria undergo the continuous process of mitochondrial network dynamics to meet the constant cellular ATP requirements i.e.,for fulfilling cellular metabolic needs and uniform distribution of mitochondria across the cell (Westermann,2012).Mitofusin 1 (MFN1),Mitofusin 2 (MFN2),and optic atrophy-1 (OPA1) proteins regulate the mitochondrial fusion process.Former 2 proteins are present on the mitochondrial outer membrane while OPA1 is present on the inner side of the mitochondrial membrane.The fission process is regulated by dynamin-related protein 1 (Drp 1) and fission 1 (Chan,2020).Abnormality in these proteins is related to excessive ROS production,synaptic dysfunction followed by synaptic loss.Increased malonaldehyde production and DNA fragmentation are the subsequent consequences of enhanced ROS.Proper mitochondrial functioning is essential for the regulation and maintenance of central nervous system (CNS) events (Yan et al.,2012).
Elevated ROS,impairment in mitochondrial dynamics,mitochondrial mutations,protein aggregation,and numerous environmental conditions may alter ATP production and metabolism in mitochondria.These changes are often linked with mitochondrial dysfunction mediated neurological disorders (Sies and Jones,2020).Figure 1
shows the possible mechanism of mitochondrial dysfunction and its link with neurological disorders.Table 3
represents a wide array of clinical signs and symptoms for these neurological disorders.However,mutations/deletions in mtDNA may be sporadic or maternally transmitted.Mitochondria have two facets that are necessary for leading a healthy life,whereas,on the other hand,it is related to neuronal cell death.Mitochondrial damage in neurological disorders has led to the tremendous development of mitochondrial-directed therapeutic strategies.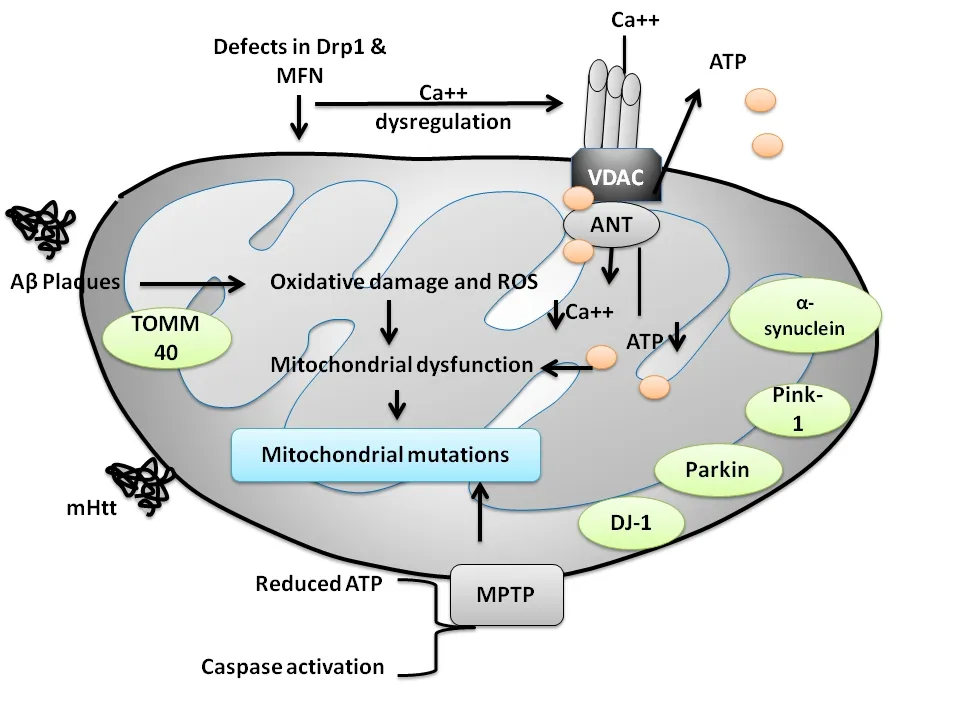
Figure 1 | Diagrammatic representation of the pathways involved in mitochondrial dysfunction in neurodegenerative disorders.
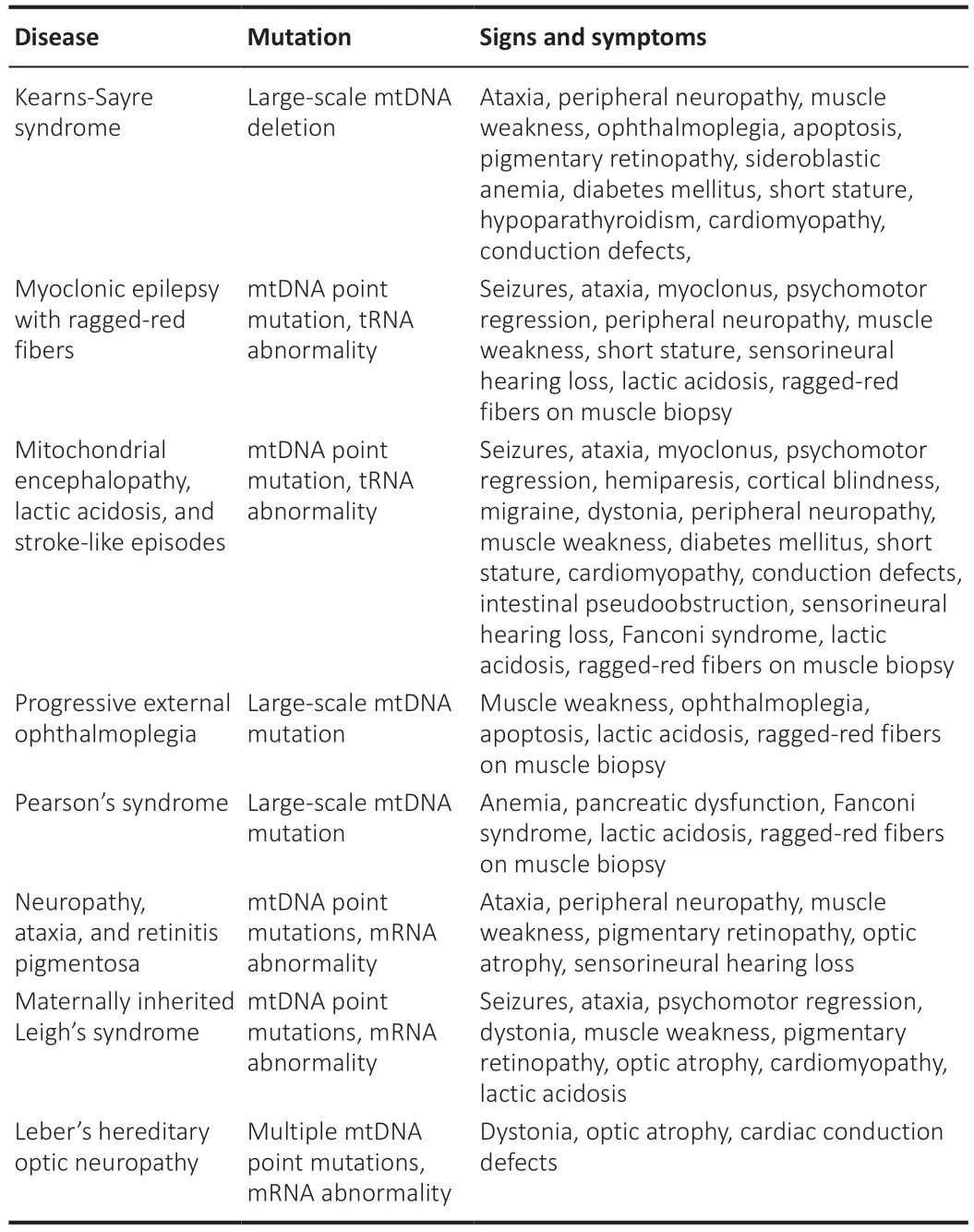
Table 3 | Signs and symptoms encountered in mitochondrial disorders
Mitochondrial Mutations and Neurological Disorders
Mitochondrial mutations are primarily linked with the changes in postmitotic cells,which exhibit increased metabolism in muscles and neurons,signifying their roles in mitochondrial disorders.Mutations can be either heteroplasmic or homoplasmic;however,most of them heteroplasmic whereas homoplasmic mutations are linked with the increased expression of mitochondrial inheritance (Pintoa and Moraesa,2014).
Alzheimer’s disease and mitochondrial alterations
Alzheimer’s disease (AD) is a complex,multifaceted disease mostly affecting people with advancing age.It is characterized by failure of acquired skills causing agnosia,aphasia,apraxia with interpersonal and social deterioration(https://www.verywellhealth.com/the-4-as-of-alzheimers-disease-98591).Dystrophic neuritis,loss of dendritic branches,and alterations in dendritic spines in AD brains are connected with the changes in mitochondria(Baloyannis,2009).Overexpression of amyloid precursor protein (APP) in transgenic mice is also coupled with the clogged mitochondrial protein import machinery.Deficient ATP production,increased ROS,cytochrome-c (COX),inhibition of complex I,and apoptosis induction are the key contributors involved in mitochondrial dysfunction (Wilkins and Swerdlow,2017).Lipid peroxidation caused by excess ROS yields toxic aldehyde causing impairment in activities of mitochondrial enzymes namely:alpha-ketoglutarate dehydrogenase and pyruvate dehydrogenase (Sies and Jones,2020).Mitochondrial damage in AD is represented through the inhibition of complex I and COX,ultimately leading to decreased cell energy and augmented tau phosphorylation causing progression of mitochondrial damage in AD (Wang et al.,2020).
Mutations in mtDNA are often correlated well with neurological impairment.Mitochondrial mutations can account for the degree,pattern,and load segregation across the whole brain as well as among each neuron in neurological disorders.Mutational modifications in the TOMM40 gene are found to be the crucial contributor and important risk factor for AD(Ferencz et al.,2012;Roses et al.,2013).The probability for AD development is facilitated by the varying poly-T 336 homopolymers in TOMM40 gene(rs10542523),which is in linkage disequilibrium 337 with APOE (Lutz et al.,2010;Roses et al.,2013).
Blood and brain tissue samples collected from experimental animal models of AD have revealed that the TOMM40 gene is a potential biomarker for AD.Microtubule-associated protein tau (MAPT) gene encodes for tau protein (Lee et al.,2012;Chong et al.,2013),even a point mutation in the MAPT gene can lead to its hyperphosphorylation,aggregation,and plaque deposition.Intriguingly,it also reduces the binding ability of tau to microtubules ultimately making it more unstable.Unstable microtubules affect axonal mitochondrial transport disrupting synaptic communication (Strang et al.,2019).Moreover,the activity of the respiratory complex,rate of fission,and fusion are decreased in hyperphosphorylated mutant tau in SY5Y cells increasing their susceptibility towards oxidative stress (Schulz et al.,2012).In brief,mutations in the MAPT gene directly affect mitochondrial function and dynamics.Evidence supports that mutations in presenilin-1 are implicated in mitochondrial dysfunction-induced AD (Hansson et al.,2004).
Mutations can be repaired by the available DNA repair pathways such as the base excision repair (BER) pathway;it is based upon the modification of the base through alkylation,deamination,and oxidation processes.Affected as well as non-affected regions of the brain represent BER deficiency,suggesting that BER impairment is common in AD brains.Diminished activities of enzymes such as uracil DNA glycosylase,OGG1,and DNA polymerase beta are responsible for causing an imbalance in the BER mechanism in the clinical condition of AD (Chatterjee and Walker,2017).The ability to repair mitochondrial nDNA is reduced in AD patients.Mitochondrial dysfunction and oxidative damage are the core contributors involved in AD pathogenesis,although the exact mechanism underlying the disease pathology is still debated (Lin and Beal,2006;Wang et al.,2020).
Parkinson’s disease
Parkinson’s disease (PD) accounts for the second most prevalent movement and widespread movement disorder.Elderly people aged over 65 years are most commonly affected by PD with a worldwide prevalence of around 2%.Degenerated dopaminergic neurons,depleted dopamine in basal ganglia,and intra-cytoplasmic inclusions containing fibrillar α-synuclein leading to the formation of Lewy bodies are some of the characteristic hallmarks of PD (Kouli et al.,2018).Genetical examination of patients with familial PD has led to the identification of numerous mutated genes such as α-synuclein (PARK1/4),parkin (PARK2),PINK1 (PARK6),DJ-1 (PARK7),LRRK2 (PARK8),and ATP13A2(PARK9),whereas more rarely associated genes include PARK3,UCHL1 (PARK5),GIGYF2 (PARK11),HTRA2 (PARK13),PLA2G6 (PARK14),and DNA polymerase G gamma (POLG) that are more often related to the prevailing risk factors for PD.However,they were not accounted for the causative mutation.Figure 2
briefly represents the associated abnormalities and mutations in mitochondrial genes and their involvement in neurodegenerative disorders(Schapira,2011).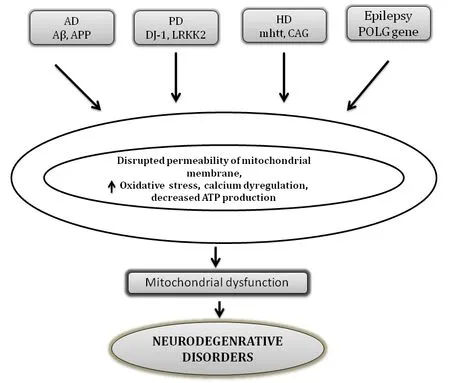
Figure 2 | Major proteins involved in mitochondrial abnormalities in various neurodegenerative disorders.
α-Synuclein
α-Synuclein (α-syn),is a presynaptic neuronal protein composed of 140 amino acid fibrillar protein that is susceptible to aggregation.The presence of water-repelling the amyloid-beta domain is an important factor,which is accountable for its aggregating property.Lewy bodies composed of a fibrillar form of α-syn are one of the major contributors involved in the pathogenesis of PD (Goedert and Spillantini,2017).Numerous studies indicate that disruption in the activity of mitochondrial respiratory complex-I,excessive mitochondrial ROS production,and reduction in mitochondrial membrane potential are some of the deteriorating effects associated with enhanced mitochondrial deposition of α-syn (Puspita et al.,2017).From the above,it is evident that α-syn accumulation and alterations in mitochondrial processes are interrelated pathological events that play a key role in α-syn-associated mitochondrial abnormalities in the progression and development of PD(Puspita et al.,2017).Genetic alterations of a single base pair (A530T,A30P,and E46K) and genomic duplications in PARK1 and SNCA region of α-syn are correlated well with an autosomal dominant PD (Polymeropoulos et al.,1997;Kruger et al.,1998;Singleton et al.,2003).Overexpression of human A53T α-syn mutation in a transgenic mouse model is linked with DNA double-strand break in neurons,and mitochondrial DNA damage in motor neurons (Martin et al.,2006).Abnormalities in mitochondria and α-syn protein accumulation are implicated in PD pathogenesis.The ability of α-syn to mediate oxidative stress may be one of the possible mechanisms involved in mitochondrial dysfunction and protein accumulation in the case of α-synuclein.An increase in activity of cytotoxic enzymes such as lactate dehydrogenase-II,carbonic anhydrase-II,the enzyme responsible for the maintenance of acid-base balance,and the increase in activity of highly abundant alpha-enolase also relate to the disease worsening and progression.Thus,modification in the activities of enzymes and proteins responsible for energy production and metabolism is highly susceptible to oxidative damage linked with mutant α-syn(Poon et al.,2005).
Parkin
Young-onset autosomal recessive Parkinson’s disease was found to be associated with the mutations in the parkin gene (PARK2) (Fang et al.,2019).Parkin localized on the outer layer of the mitochondrial surface diminishes the release of COX and inhibits mitochondrial swelling playing a key role in guarding cells against cellular apoptosis (Zhang et al.,2016).The protective role of parkin has been demonstrated in parkin knock out mouse and fly models,which illustrates that mice and flies lacking parkin are more prone to develop mitochondrial abnormalities such as mitochondrial blebbing,disintegrated cristae,and the decline in activities of complex I and IV of the mitochondrial respiratory chain (Shim et al.,2011;Steele et al.,2014;Fichi et al.,2019).Contrary to this,mice overexpressing parkin observed reduced dopaminergic cell loss in the MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) model of PD.These protective responses confer mitochondrial defense and decrease in α-syn.Parkin is necessary for the maintenance and regulation of mitochondrial dynamics and autophagy (Bian et al.,2012;Sun et al.,2012).
Phosphatase and tensin homolog-induced putative kinase 1
Phosphatase and tensin homolog-induced putative kinase 1 (PINK1),a protein kinase localized to mitochondrial membranes,is predominantly expressed in neurons of the human brain.After parkin,genetic alterations linked with PINK1 are the second most prominent cause contributing to the development and progression of early-onset autosomal recessive PD.Mitochondrial abnormalities noted in the PINK1 knock-out mouse model are quite similar to those noted in parkin such as decreased mitochondrial membrane potential,structural changes in mitochondria,increased activity of caspase,and enhanced oxidative load (Gonçalves and Morais,2021).Furthermore,mitochondrial proteins in the midbrain,striatum,and cerebral cortex are responsible for regulating metabolic hemostasis and mitochondrial membrane potential also undergo alteration in the disease progression.In terms of specificity,substantia nigra in the midbrain region was severely affected and exhibits a maximum number of defective mitochondria.Furthermore,alterations in these mitochondrial proteins are connected with the mutations in PINK1.Mutations in PINK1 induce structural changes in mitochondria and serve as an important mediator of ROS production (Kausar et al.,2018).
Protein deglycase DJ-1 and leucine-rich repeat kinase 2
Mutations in protein deglycase DJ-1 and leucine-rich repeat kinase 2 (LRRK2)are indicated in the sporadic form of PD.Approximately 1–2% of cases of early-onset PD are associated with DJ-1,(Repici and Giorgini,2019),whereas LRRK2 is known to induce autosomal-dominant cases of PD.The majority of the cases associated with DJ-1 and LRRK2 are related to familial cases of PD,with less frequent cases for the occurrence of sporadic late-onset PD (Kessler et al,2018;Repici and Giorgini,2019).Several lines of experimental evidence indicate that knockdown or overexpression of DJ-1 directly or indirectly corresponds to increased susceptibility for cellular death.Though DJ-1 possesses the capability to escape cellular death owing to its cytoprotective potential;however,its cytoprotective potential is limited to prevent oxidative stress-induced cell death,and it does not seem to confer protection against cell death occurring due to any other causes (Mihoub et al.,2017;Raninga et al.,2017).Cytoprotective property is subjected to the alterations in cysteine residues of DJ-1.Under the conditions of extreme oxidative stress,cysteine residues undergo modification to either cysteine-sulfinic or cysteinesulfonic acid,or both.Interestingly,DJ-1 has also been reported to exhibit neuroprotective effects possibly by acting as transcriptional co-activator protease or a molecular chaperone (Takahashi-Niki et al.,2017).
Knowledge about the physiological function of LRRK2 protein is limited,but as mentioned above,it is involved in the progression and development of autosomal recessive dominant cases of PD.The precise physiological role of LRRK2 in this protein is unidentified,but the presence of multiple functional domains suggests involvement in a wide array of functions.A possible site of action for the LRRK2 mechanism is mitochondria.A brief update about mitochondrial DNA damage in neurological disorders is shown in Table 4
(West et al.,2005).
Table 4 | Indication for the participation of mitochondrial DNA damage in neurological disorders
Evidence from subcellular fractionation studies has established a fact that 10% of total LRRK2 protein is coupled with the mitochondrial fraction.Immunohistochemical results further support the localization of LRRK2 to mitochondria.According to previous reports,LRRK2 also shares a cross-talk with TOM20 (Biskup et al.,2006;Gloeckner et al.,2006).This finding was further confirmed by repurposing polyclonal antibodies to amino or carboxyl terminus of LRRK2 to investigate the subcellular localization of LRRK2 in microsomal,synaptic vesicle-enriched,and synaptosomal fractions of rat brain slices and mitochondria.
POLG (mitochondrial DNA polymerase gamma gene)
An earlier report suggests that mitochondrial dysfunction holds significant relevance in pathogenetic forms of PD,specifically sporadic and idiopathic forms (Schapira et al.,2011).Evidence indicates that nuclear-encoded protein(POLG) is implicated in the production,replication,and repairing of mtDNA.Any mutation in the catalytic subunit of PLOG is connected with the depletion or deletions of mtDNA in several types of neurological phenotypes including PD (Stiles et al.,2016;Young and Copeland,2016).These properties of the catalytic subunit are attributed to its polymerase activity at 59 to 39 and exonuclease activity at 39 to 59.Evidence indicates that clustering of somatic mtDNA deletions occurs in the substantia nigra in PD cases.Moreover,gene products of familial Parkinsonism such as POLG1 have a crosslinking with mitochondrial function.Several lines of evidence explicit that mutations in POLG1 are associated with levodopa-induced Parkinsonism in some families(Hudson et al.,2007;Luoma et al.,2007;Eerola et al.,2010).Mutations in POLG1 lead to the incorporation of defective or incorrect nucleotides in mtDNA causing impairment in the mitochondrial respiratory chain (Orsucci et al.,2011).One report illustrates that homozygous mice having polymerase deficient mutant of POLG1 express a phenotype with enhanced point mutations and mtDNA deletions (Trifunovic et al.,2004).In a few cases,genetic abnormalities occurring in mtDNA due to POLG1 gene mutation can directly or indirectly cause Parkinsonism (Luoma et al.,2007;Eerola et al.,2010).Missense mutations in POLG1 such as a mutation in G737R and R853W are represented in some cases of early-onset Parkinsonism (Davidzon et al.,2006).
Huntington’s disease
Huntington’s disease (HD) is an autosomal dominant neurodegenerative movement disorder.HD is characterized by degeneration of striatal GABAergic medium spiny neurons leading to the development of motor and nonmotor symptoms viz involuntary muscle movements,psychiatric disorders,and memory loss (https://www.ninds.nih.gov/Disorders/All-Disorders/Huntingtons-Disease-Information-Page;accessed on October 8,2021).Mitochondrial defects in HD are attributed to the defects and mutations in the huntingtin (htt) gene and CAG triplet repeat expansion.CAG triplet repeat expansion is responsible for generating a polyglutamine (polyQ) stretch inmutant
htt
(mhtt
) promoting its accumulation in the brain (Neueder et al.,2017).Themhtt
produces alterations in ATP production and consumption,mhtt
and various other vital cellular processes including endocytosis,intraneuronal trafficking,transcriptional regulation,postsynaptic signaling,and initiation of the apoptotic cascade,probably via causing alteration in mitochondrial network dynamics (Song et al.,2011;Wang et al.,2013;Guo et al.,2016).Another serine protease protein HtrA Serine Peptidase 2 (HTRA2)has been reported to be present on the inter-mitochondrial membrane.HTRA2 known to induce HD-like phenotypical modifications in transgenic mice is located in the inter-mitochondrial membrane.HTRA2 levels were reduced in striatal neurons in conditions wheremhtt
is expressed.Crosstalk between HTRA2 andmhtt
indicates HTRA2 homeostasis is coupled with the specific susceptibility to the striatal neurons (Inagaki et al.,2008).Apart from HTRA2,peroxisome proliferator-activated receptor-g coactivator 1α (PGC-1α)is one of the critical targets,which is involved in the regulation of a few other metabolic processes such as OXPHOS,mitochondrial biogenesis,and adaptive thermogenesis has also been reported (Hollenbeck et al.,2005;Lin et al.,2005;Puigserver et al.,2005;Cui et al.,2006;).Epilepsy
Epilepsy shares a widespread prevalence of 0.5–0.7% across the globe.It is characterized by episodes of recurrent seizures,synchronized neuronal discharges affecting normal brain functioning.Epileptic seizures present the signs of mitochondrial abnormalities and disparities from normal behavior in the CNS.Mitochondria are the power house of the cells,responsible for ATP production,which is vital for neuronal excitability and survival (Singh et al.,2020).In neurons,balanced mitochondrial functioning is utilized to carry out physiological processes such as neurons Cahemostasis,redox machinery,developmental and synaptic plasticity,and cell cycle (Singh et al.,2020).High energy demand and lack of regenerative capacity of CNS neurons increase the likelihood of CNS towards mitochondrial dysfunction and increased frequency of occurring epileptic seizure.In response to defects in the mitochondrial respiratory chain,there is a decreased intracellular ATP level,increased neuronal excitability,impairment in Na/KATPase activity,and reduced mitochondrial membrane potential (Vezzani et al.,2019).
Energy depletion in mitochondria is correlated well with enhanced synaptosomal and astrocytic glutamate release indicating impairment in the glutamate-aspartate transporter in mitochondria (Vezzani et al.,2019;Singh et al.,2020).Figure 3
illustrates the basic mechanism involved in the occurrence of epileptic seizures.Genetic alterations in mtDNA and nuclear genes can also lead to epilepsy.Mitochondrial encephalopathy,lactic acidosis,and strokelike episodes (MELAS);MERRF,myoclonic epilepsy,and ragged-red fibers(MERRF) are the two disorders that are linked to the mutations in mtDNA.m.8344A>G mutation in the mitochondrial tRNA gene for the lysine gene,MTTK,was the first-ever reported mutation (Hou et al.,2020).m.8356T>C and m.8361G>A,are other two mutations,which are known to cause the same clinical condition.The first incidence of MELAS came to light in 1984,affecting 1:15,000 individuals worldwide with varying phenotypic expression (Bhatti et al.,2017;Zhu et al.,2018).Maternally inherited neurodegenerative disorder MELAS occurs due to mutation in m.3243A>G.Nearly 80% cases of MELAS are associated with the mutation m.3243A>G of the mitochondrial transfer RNA(tRNA) (Leu (UUR)) gene (MT-TL1) (Montagna et al.,1988;Goto et al.,1990).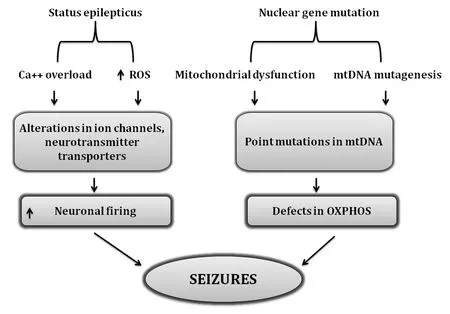
Figure 3 | Association of mutational modifications and ROS-related cascade in deterioration of epileptic seizures.
Therapeutic Strategies Targeting Mitochondrial Dysfunction
Mitochondrial dysfunction is implicated in the pathophysiology of neurological disorders.Alterations in OXPHOS,enhanced mtDNA deletions,mutations,deregulation in calcium signaling,and impairment in energy metabolism including crosstalk with pathogenic target proteins (e.g.,amyloid-β,α-syn,parkin,PINK1,and huntingtin) are the major factors affecting mitochondrial function and biogenesis.It is noteworthy to state that mitochondrial pathology could be potentially involved in the onset of clinical presentation of AD,PD,HD,and epilepsy kind of neurological disorders.Thus,therapeutic interventions targeting to strengthen mitochondrial functions would represent an attractive strategy to improve the pathological alterations observed in these disorders.Therapeutic approaches targeting mitochondrial dysfunction in neurological disorders are multifaceted.Antioxidants,compounds targeting mitochondria such as alpha-lipoic acid,coenzyme Q10,carnitine,vitamin C,vitamin E,methylene blue,Ginkgo biloba,piracetam,simvastatin,curcumin,and omega-3 polyunsaturated fatty acids are some of the conventional strategies,which are proven to be beneficial (Zhao and Fu,2021).Current strategies are focusing on proper regulation of (i) mitochondrial fusion/fission process(ii) mitochondrial proteins (iii) gene therapy to decrease mutant mtDNA (iv)peptides and lipophilic cations targeting mitochondria,might represent a novel therapeutic strategy.Recently,newer perspectives in mitochondrial therapeutics are the modulation of mitochondrial network dynamics.Improvement in mitochondrial dynamics is achieved by mitochondrial biogenesis stimulating mitochondrial movement,and nucleotide supplementation (Leitão Rocha et al.,2015).
Drugs targeting increased mitochondrial fusion/fission process
Emphasis has been laid down towards developing suitable and safer drug candidates aimed at reducing the enhanced mitochondrial fission/fusion processes.These approaches are aimed at maintaining proper balance in mitochondrial network dynamics within the mitochondria.It has been hypothesized that the development of these novel strategies may exert some beneficial actions in upgrading mitochondrial functioning,neuronal activity,and cell survival in neurological disorders associated with oxidative stress and mitochondrial dysfunction.To date,three inhibitors for improving mitochondrial fission are screened through different chemical libraries.Mdivi (Cassidy-Stone,2008),Dynasore (Macia et al.,2006),and P110 (Qi et al.,2013) are the available inhibitors for correcting impairment in the mitochondrial fission process.
Mdivi
A considerable amount of work has been done on the Mdivi molecule in experimental rodent models for epilepsy and/or seizures for exploring its protective potential (Kim et al.,2016).Qi et al.(2013) have demonstrated that in anin vivo
study,Mdivi-1 can inhibit mitochondrial fission in an epileptic rat model.Further,the findings of their study reveal that seizures can cause an increment in the mitochondrial fission and contrary mdivi-1 treatment significantly attenuated oxidative damage with the considerable improvement in neuronal density in the hippocampus.Mdivi also acts as a ROS scavenger against oxidative damage,mitochondrial toxicity ischemia (Qi et al.,2013),oxygen-glucose deprivation (Tang et al.,2013),and rhabdomyolysis (Chlystun et al.,2013).Proven beneficial effects of Mdivi have made it an excellent drug of choice for combating and inhibiting the disproportionate mitochondrial fission and for maintenance of fission-fusion balance normal and the normal functioning of cells.Dynasore
Dynasore has gained significant relevance as a dynamin GTPase inhibitor for endocytic pathways.It prevents fragmentation and helps in making dynamindependent endocytic vesicles.Over 16,000 small molecules were screened by Macìa et al.(2006) to identify their potential to inhibit the mitochondrial fission process.During screening novel compounds,they found a new compound and named it“dynasore”.Due to its remarkable property of inhibiting the fission,dynasore is considered to bring notable changes in the mitochondria-targeted approaches,which claim to balance the mitochondrial fission.Therefore,more stability is provided in mitochondrial network dynamics (Macia et al.,2006).
P110
The development of P110 is another successful strategy under the category of fission inhibitors.Initially,P110 was found to be efficient in blocking the enzyme activity of Drp1,decreasing Fis1 showing improvement in mitochondrial fissionin vitro
.P110 was further explored for various properties.The evidence further indicates the neuroprotective potential of P110 probably via its inhibitory action on mitochondrial fission and ROS generation.Inhibitory action of P110 on mitochondrial fission is responsible for maintaining sustained mitochondrial integrity and membrane potential.Nevertheless,an additional research is needed to raise the curtain for disclosing its potency as an efficient Drp1 inhibitor (Qi et al.,2013).Therefore,Mdivi,Dynasore,and P110 represent an interesting strategy to treat neurological disorders that deal with disturbances in mitochondrial fission and dysfunction.
Drugs targeting mitochondrial proteins
Targeting mitochondrial proteins is another fascinating scheme that might represent a unique therapeutic approach directed towards these neurological disorders.Presently,significant attention is being given to developing various mitochondria-directed antioxidants to treat and improve mitochondrial dysfunction.Antioxidant therapy is divided into three main categories:(i)Stoichiometric scavengers:nitroxide spin traps,vitamin E or C (ii) catalytic antioxidants:salen,or metalloporphyrin’s,and lastly (iii) indirect antioxidants:iron chelators.Activation of Nrf2 pathway is the basic mechanism for obtaining the desired protective effects linked with iron chelators,and it also activates various anti-oxidant pathways primarily Nrf2,increasing mitochondrial GSH,and decreasing ROS production (Csire et al.,2020).Vitamin E is a wellknown antioxidant that confers neuroprotection against pilocarpine-induced epilepsy.Moreover,these studies have concluded vitamin E can significantly increase the catalase and free fatty acid in the brain.A previous report further indicates the beneficial effects of vitamin E co-medication in improving the cases of refractory epilepsy by substantially reducing the occurrence of seizure frequency in child patients (Mehvari et al.,2016).Although clinical trials with vitamin E are still controversial having numerous unsuccessful attempts for decreasing the occurrence of seizures in pediatric patients and epilepsy animal models (Levy et al.,1992;Xu and Stringer,2008).Numerousin vitro
andin vivo
studies indicate the protective potential of taurine against mitochondrial-induced damage and related neurological disorder such as MELAS (Homma et al.,2021).Decreased frequency of stroke-like episodes and alterations in mitochondrial tRNAis also reported with oral administration of taurine in MELAS patients (Ohsawa et al.,2019).Preventive effects of beta-alanine induced mitochondrial damage,drop in mitochondrial membrane potential,and improvement in apoptosis were also noted in mouse embryonic fibroblasts treated with beta-alanine (Shetewy et al.,2016),which showed that taurine pretreatment protects against mitochondria damage and mitochondria fission in beta-alanine-treated mouse embryonic fibroblasts (Jong et al.,2012).The antioxidant potential of taurine is related to its ability to scavenge hypochlorous acid (HOCl),produced from hydrogen peroxide (HO) during ROS generation (Li et al.,2004).This property of taurine is utilized for its ability to prevent mito-oxidative stress,sustenance of intracellular calcium homeostasis against oxidative insult in mitochondria (El Idrissi and Trenkner,2004;El Idrissi et al.,2008).Gene therapy to reduce mutant mtDNA
mtDNA is heteroplasmic.To combat this disease condition gene-based therapeutic interventions are considered to be an important approach,which claims its preventive action by maintaining an equilibrium towards wildtype mtDNA by declining the mutant mtDNA below the threshold of these neurological disorders.These strategies are involved in degrading the mutant mtDNA by selective mitochondrial nucleases (Smith and Lightowlers,2011;Leitão-Rocha et al,2015).In this context,anti-replicative therapy is one such strategy involving sequence-specific nucleic acids designed to bind mtDNA replication,promoting the propagation and replication of wild-type mtDNA.Peptide nucleic acid has shown beneficial effectsin vitro
;however,it lacks to modify heteroplasmy under a condition where cells are in intact form due to the presence of an increased amount of mt DNA (Smith and Lightowlers,2011;Leitão-Rocha et al,2015).Current reports suggest that tRNA’s possess the ability to deliver antireplicative oligo-ribonucleotides to mitochondria through RNA import pathways.tRNAs have been shown to reduce the mtDNA load by 15–35%in Kearns-Sayre syndrome (Comte et al.,2013).Moreover,endonucleasemediated heteroplasmy is also a newer approach for mutant mtDNA.
Peptides and lipophilic cations targeting mitochondria
The inability of mitochondria-targeted antioxidants to increase the antioxidant levels within mitochondria has posed a serious limitation to its use.At present much enthusiasm has been delivered to innovate and develop mitochondriaspecific antioxidant therapies (Armstrong,2007).Conjugation of lipophilic triphenylphosphonium (TPP) cation to an antioxidant moiety,like coenzyme Q (MitCoQ) and α-tocopherol (MitoVitE) are the newer approaches in this track (Murphy,2000).Potential gradient across the mitochondrial inner membrane plays an important role in generating the efficacy of lipophilic cation.Consequently,the proton gradient produces a negative potential of 150 to 180 mV across the inner membrane causing the lipophilic cations to accumulate at 100 to 1000 times in mitochondria.Similarly,MitoVitE is occupied by mitochondria approx.80-fold more than vitamin E,and considered as far more effective in defending mitochondria against oxidative load as vitamin E itself.
MitoVit E is also found to be 800 times more effective than idebenone,a CoQ10 analog in imparting protection against glutathione depletion in fibroblast cultures obtained from patients with Friedreich ataxia and it is highly approx 350 times more efficacious than trolox,another vitamin E analog (Suárez-Rivero,2021).Current strategies are now also aimed at developing mitochondria-directed peptides:Szeto-Schiller (SS) peptides(Szeto,2006).Some analogs of SS peptides are discussed here in the present review.Peptide SS-31 has a significant potency which can be related well by its extensive cellular uptake and distinguished partitioning into mitochondria.Moreover,[3H] SS-31 has high intracellular concentrations,6-fold higher than extracellular concentrations.Drastic changes in [3H] SS-31 concentration have also been noted in the isolated mitochondrion,which reveals that it was concentrated to about 5000-fold in the mitochondrial fraction.By accumulating in the inner mitochondrial membrane,SS-31 localizes at the site of ROS production thus creating hindrance for ROS mediated damage conferring protection against mito-oxidative damage and further ROS production (Misrani et al.,2021).Reports also provide support to the fact that SS-31 shields neuronal cells against tert-butyl-hydroperoxide caused mitochondrial depolarization and programmed cell death (apoptosis) by declining intracellular ROS levels and caspase activity (Zhao et al.,2005).It reduces the mitochondrial generation of ROS and inhibits PTP and depolarization in isolated mitochondria (Misrani et al.,2004).Recently,Yang et al.(2009) examined the properties of SS-31 and SS-20 to defend against MPTP-induced neurotoxicity in mice.
Emerging Perspectives in Therapeutics
Mitochondrial biogenesis
Mitochondrial diseases are characterized by defective mitochondrial network dynamics i.e.,bioenergetics defects.Therefore,therapeutic approaches for improving cellular ATP production would represent an exciting strategy.Peroxisome proliferator-activate receptor (PPAR) and PGC-1α,one of its coactivators are major contributors in initiating biogenesis,demonstrating PPAR/PGCtrα as an important drug target.It has been reported that transgenic mice expressing PGC-1α,or treatment with bezafibrate (PPAR agonist)induces mitochondrial biogenesis,prevents mitochondrial myopathy in mice having deficient COX activity in skeletal muscle.However,treatment with bezafibrate failed to increase ATP production via mitochondrial biogenesis in COX efficient animals though it shows the improvement in PGCgh expression and preventing disease conditions.In a mouse model of encephalopathy,bezafibrate exhibits the neuroprotective potential in mitochondrial encephalopathy (Dumont et al.,2012;Noe et al.,2013).Lysine deacetylases(KDACs) regulate mitochondrial biogenesis through altering the activity of PGCrou gene expression implying that KDAC modulatory drugs represent a successful strategy in improving mitochondrial biogenesis (Guedesesisr and Oliveira,2013).
Movement and mitophagy
Activation of mitochondrial movement is a promising scheme to rule over the abnormalities in mitochondrial distribution as some neurons demand extensive energy and so mitochondria must travel those distant synapses.In AD and HD brains,tubulin acetylation is significantly reduced;disturbances in microtubule-dependent mitochondrial transport are evident by their cellular models (Dan et al.,2018).Microtubule deacetylase inhibitors such as HDAC6 in conjunction with tubacin have been reported to augment motor recruitment,promoting two-directional mitochondrial transport in cellular models of HD.One more HDAC6 inhibitor tuba statin A has shown beneficial effects in mitochondrial transport in hippocampal neurons of amyloid-β induced AD rats (Dompierre et al.,2007;Govindarajan et al.,2013).
Mitophagy dysregulation is associated with neurodegeneration and can be either defective or excessive (Wanderoy et al.,2021). In the case of PD,the PINK1/parkin pathway has emerged as a major contributor to mammalian mitophagy.Disrupted mitochondria with distorted ∆ψm cannot import and process PINK1,causing its accumulation outside the mitochondria recruiting the ubiquitin-ligase Parkin.Ubiquitinated mitochondria are identified by ubiquitin-binding adaptors like p62 and HDAC6.This recognition process facilitates its interaction with the autophagosomal protein.Microtubuleassociated protein 1A/1B-light chain 3 is a soluble protein that establishes a cascade that defines the mitochondrial digestion and functioning of autophagosome-lysosome fusion (Quinn et al.,2020).Presently,the (patho)physiological factors that determine neuronal mitophagy and the mechanisms for treating injured mitochondria in isolated synapses are poorly understood presenting a terrific challenge for the discovery and development of mitophagy-associated therapeutic strategies.
Nucleotide supplementation
Advancements have been made for treating imbalance in mitochondrial functioning starting from its dysfunction to mtDNA depletion.Deoxyribonucleotides supplements are an emerging perspective designed to improve mtDNA depletion in fibroblasts of patients having mutations in enzymes associated with the regulation and control of mitochondrial nucleotide and deoxynucleotide pools.The most common enzymes linked with this function are deoxy-guanosine kinase and thymidine phosphorylase(TYMP).Deoxyguanosine kinase DGUOK and TYMP genes encode TP.In a TP knockout mouse,mtDNA depletion has been modified by dCtd or tetrahydro uridine treatment,a nucleotide catabolism inhibitor (Camara et al.,2014).TYMP1 mutations encoding TP are related to the onset of mitochondrial neuro-gastro-intestinal encephalomyopathy (Nishino et al.,1999).
Conclusion
Oxidative stress,mitochondrial dysfunction,and mitochondrial mutations have interrelated to the pathogenesis of neurological disorders.Increased oxidative load augments ROS production leading to dysfunction and mutations in mitochondria.Mitochondria are the power house of the cell that regulates important aspects of cellular functions in a wide variety of cell organelles such as the nucleus and endoplasmic reticulum mutations.Mutations in mtDNA genome may hamper the proper functioning of mitochondria and cellular functions causing perturbations in mitochondrial stability eliciting a series of changes in nuclear gene expression.Therefore,it is of significant interest to explore and examine the mitochondrial mutations involved in the progression,development,and worsening of neurological disorders.Further,identification of these mutations and associated changes may also open new horizons for the development of therapeutic strategies aimed at improving these conditions.Nevertheless,the precise mechanisms underlying these abnormalities and mutations in mitochondria need to be explored.Some of the achievements done so far concerning identification of treatment strategies include molecular approaches such as (i) targeted reexpression of the mutated gene (ii) manipulating mtDNA heteroplasmy (iii)stabilizing mutant mt-tRNA (iv) bypassing the block of the respiratory chain are in pipeline for establishing their role in improving these abnormalities.Identification of mutations in mitochondria could be an attractive approach for the development of novel drugs aimed to combat the abnormalities in mitochondria and associated alterations which might have resulted due to these mutations.
Acknowledgments:
I express my sincere gratitude towards my mentors Dr.Atish Prakash (Scientist,Eurofins PSS Insourcing solutions,Malvern,PA,USA),Dr.Puneet Kumar Bansal (Associate Professor of Pharmacology &Head,Department of Pharmacology,Central University of Punjab,Bathinda,Punjab,India),and Dr.Avril V.Somlyo (Professor,Department of Molecular Physiology and Biological Physics,School of Medicine,University of Virginia,Charlottesville,VA,USA) for their guidance and valuable inputs.I am thankful to Mr.Praveen Garg,Chairman,ISF College of Pharmacy,Moga,Punjab,India for his continuous support and encouragement.
Author contributions:
Design,concept,idea,manuscript writing,data search and all the changes made till the final manuscript were done by JK.The author approved the final version of the manuscript.
Conflicts of interest:
The author declares no conflicts of interest.
Open access statement:
This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.

- 中国神经再生研究(英文版)的其它文章
- Neuroaxonal and cellular damage/protection by prostanoid receptor ligands,fatty acid derivatives and associated enzyme inhibitors
- Extracellular vesicles in Alzheimer’s disease:from pathology to therapeutic approaches
- Molecular approaches for spinal cord injury treatment
- Sex-biased autophagy as a potential mechanism mediating sex differences in ischemic stroke outcome
- Adipose tissue,systematic inflammation,and neurodegenerative diseases
- Interleukin-1:an important target for perinatal neuroprotection?