
Regulatory mechanisms of retinal ganglion cell death in normal tension glaucoma and potential therapies
2022-05-25 09:26WenCuiShenBingQingHuangJinYang
中国神经再生研究(英文版) 2023年1期
Wen-Cui Shen ,Bing-Qing Huang ,Jin Yang,
Abstract Normal tension glaucoma (NTG) is a multifactorial optic neuropathy characterized by normal intraocular pressure,progressive retinal ganglion cell (RGC) death,and glaucomatous visual field loss.Recent studies have described the mechanisms underlying the pathogenesis of NTG.In addition to controlling intraocular pressure,neuroprotection and reduction of RGC degeneration may be beneficial therapies for NTG.In this review,we summarized the main regulatory mechanisms of RGC death in NTG,including autophagy,glutamate neurotoxicity,oxidative stress,neuroinflammation,immunity,and vasoconstriction.Autophagy can be induced by retinal hypoxia and axonal damage.In this process,ischemia can cause mutations of optineurin and activate the nuclear factor-kappa B pathway.Glutamate neurotoxicity is induced by the over-stimulation of N-methyl-D-aspartate membrane receptors by glutamate,which occurs in RGCs and induces progressive glaucomatous optic neuropathy.Oxidative stress also participates in NTG-related glaucomatous optic neuropathy.It impairs the mitochondrial and DNA function of RGCs through the apoptosis signal-regulating kinase-JUN N-terminal kinase pathway.Moreover,it increases inflammation and the immune response of RGCs.Endothelin 1 causes endothelial dysfunction and impairment of ocular blood flow,promoting vasospasm and glaucomatous optic neuropathy,as a result of NTG.In conclusion,we discussed research progress on potential options for the protection of RGCs,including TANK binding kinase 1 inhibitors regulating autophagy,N-methyl-D-aspartate receptor antagonists inhibiting glutamate toxicity,ASK1 inhibitors regulating mitochondrial function,and antioxidants inhibiting oxidative stress.In NTG,RGC death is regulated by a network of mechanisms,while various potential targets protect RGCs.Collectively,these findings provide insight into the pathogenesis of NTG and potential therapeutic strategies.
Key Words:autophagy;endothelin 1;glutamate neurotoxicity;inhibitor;nerve regeneration;neuroinflammation;normal tension glaucoma;oxidative stress;retinal ganglion cell;vasoconstriction
Introduction
Glaucoma is the second leading cause of blindness globally,and is expected to affect 118 million individuals by 2040 (Tham et al.,2014).Normal tension glaucoma (NTG) is a type of open-angle glaucoma with normal intraocular pressure (IOP) (i.e.,≤ 21 mmHg in 24 hours without medication) (Hirooka et al.,2021).NTG is a type of progressive glaucomatous optic neuropathy (GON)with higher prevalence in Asiaversus
other geographic regions (Esporcatte and Tavares,2016).In NTG,optic disc excavation,retinal ganglion cell (RGC)death,and visual field defects are detected despite an IOP within the normal range (Cho and Kee,2014).It has been reported that the IOP of patients with NTG often fluctuates between 15 and 20 mmHg (Kosior-Jarecka et al.,2016b).The body of RGCs is located in the retina,with their axons projecting to the brain nuclei through the optic nerve (Harada et al.,2020).The mechanism through which RGCs respond to injury has been investigated in various models(Daniel et al.,2018).The concept of neuroprotection against RGC death in glaucoma has been attracting considerable attention (Levin and Peeples,2008;Francesca et al.,2015;Gossman et al.,2016).A previous study reported that vascular factors are involved in the pathogenesis of NTG;moreover,oxidative stress,vasospasm,and endothelial dysfunction were identified as risk factors for the development of GON(Fan et al.,2015).Moreover,Trivli et al.(2019) summarized the vascular,mechanical,and genetic mechanisms involved in the pathogenesis of NTG.The prevention of RGC death and neuroprotection are important in NTG.Therefore,the objective of this review was to discuss the mechanisms involved in the regulation of RGC death,including related signaling pathways and transcription factors,as well as emphasize potential therapies for NTG.
Search Strategy
Studies on the mechanisms regulating RGC death in NTG,published from 1989 to 2022,were retrieved from the PubMed database.The search was conducted using the following terms:“autophagy”AND“RGC”OR“NTG”OR“normal tension glaucoma”;“glutamate”AND“RGC”OR“NTG”OR“normal tension glaucoma”;“oxidative stress”AND“RGC”OR“NTG”OR“normal tension glaucoma”;“neuroinflammation”AND“RGC”OR“NTG”OR“normal tension glaucoma”;“vasoconstriction”AND“RGC”OR“NTG”OR“normal tension glaucoma”.The results of this search were further evaluated by screening the titles and abstracts of the articles.
Autophagy
Role of autophagy in RGCs
Autophagy,a highly conserved metabolic process,involves the degradation and recycling of cellular components (Klionsky,2007).It participates in both cell death and survival (Maiese et al.,2012),which can be initiated by various stress signals,such as hypoxia,ionizing radiation,infections,and chemotherapeutic agents (Cadwell and Ken,2016).Several autophagyrelated phases are involved in this process:initiation/nucleation,elongation,maturation,fusion,and degradation (Fernández-Albarral et al.,2021).Initiation and nucleation are induced through nutrient starvation,during which an insulating membrane that engulfs damaged proteins and organelles is formed.Subsequently,the phagophore extends into a mature and closed autophagosome;these phases are termed elongation and maturation.Finally,in the fusion and degradation phases,the mature autophagosome fuses with a lysosome to form an autolysosome,followed by the degradation of its contents by proteases and lipases.
The process of autophagy is regulated by numerous factors,and the normal cellular and tissue homeostasis depends on well-regulated autophagy (Cao et al.,2014).Previous studies have revealed that the autophagic capacity of cells plays a vital role in inflammatory and neurodegenerative diseases (Cuervo et al.,2005;Huang and Klionsky,2007;Levine and Kroemer,2008;Martinez et al.,2016;Jiang et al.,2022;Rickman et al.,2022).Therefore,the process of autophagy is closely related to the pathogenesis of several major ocular diseases (Kim et al.,2010;Chen et al.,2013;Dammak et al.,2021).
Autophagy can be initiated by both retinal hypoxia and axonal damage(Sase et al.,2020;Li et al.,2021;Tang et al.,2021).In the dendrites of RGCs,the process of autophagy is launched to promote cellular protection (Lin and Kuang,2014).In vitro
andin vivo
studies revealed that the autophagy pathway promotes RGC neuroprotection,and autophagy may be an effective therapeutic method for inducing regeneration of the central nervous system(CNS) (Beckers et al.,2021;Mázala-de-Oliveira et al.,2022).Inhibition of autophagy induces the accumulation of apoptotic cells in the retinal neuroepithelium,where the proliferating neuroblasts differentiate into RGCs(Mellén et al.,2008).However,accelerating autophagy will aggravate the degradation of axons in traumatically injured RGCs (Figure 1
).Heterozygous mutations of optineurin (OPTN) have been reported in some familial and sporadic cases of NTG (Fuse et al.,2004;He et al.,2019).The Glu50Lys (E50K) mutation of OPTN in Caucasian and Hispanic populations has been associated with the progression of NTG (Tin et al.,2005).However,the Met98Lys (M98K) mutation was more commonly observed in Asiansversus
other populations (Alward et al.,2003).It has been verified that even transient expression of E50K-mutated OPTN can induce apoptosis and death of RGC-5 cells (Chalasani et al.,2007).An initial study showed that homozygous OPTN knockout (KO) mice had reduced visual deterioration and preserved visual function,further supporting that suppression of OPTN may constitute a therapeutic strategy against glaucomatous neurodegeneration in NTG (Adi et al.,2021).The humanOPTN
gene codes for a 577-amino acid protein;it is organized into various domains,such as the nuclear factorkappa B (NF-κB) essential molecule (NEMO)-like domain,ubiquitin-binding domain,leucine-zipper domain,and microtubule-associated protein 1 light chain 3 (Rezaie et al.,2002;Ying and Yue,2012).As an autophagy receptor,OPTN plays an important role in autophagy,involving the degradation of ubiquitinated protein,mitochondrial damage,and bacterial ubiquitination(Korac et al.,2013;Wong and Holzbaur,2014).Moreover,OPTN is a negative regulator of the NF-κB pathway,which is also linked to autophagy.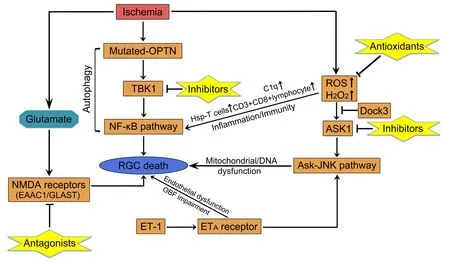
Figure 1 | Regulatory mechanisms of RGC death and potential therapeutic targets for NTG.
A close interaction between TANK (TRAF family member associated NF-κB activator)-binding kinase 1 (TBK1) and mutant E50K OPTN protein,which protects mutant OPTN from dissolution,has been reported (Yuriko et al.,2013).TBK1 is a serine or threonine protein kinase,which is a non-canonical member of the IkB kinase (IKK) family and mediates the ability of TANK to initiate the NF-κB pathway (Pomerantz and Baltimore,1999).TBK1 also participates in autophagy through phosphorylation of autophagy receptor proteins (Richter et al.,2016;Durand et al.,2018).In pedigrees with NTG,several research studies discovered copy number variations spanning theTBK1
gene,including duplications and deletion (Ritch et al.,2014;Awadalla et al.,2015;Kaurani et al.,2016).Particularly,mutations in both OPTN and TBK1 are closely related to 1–2% of NTG cases (Fingert,2011).In a recent study,syntaxin 17,which has been implicated in autophagosome-lysosome fusion,was found to be a substrate for TBK1 phosphorylation.In summary,both syntaxin 17 and TBK1 play a vital role in the initiation of autophagy (Kumar et al.,2019).Small molecules and therapeutic potential
TBK1 and IKK inhibitors have been developed and investigated in some cellbased studies and animal models (Table 1
).Amlexanox,a specific small molecule inhibitor of TBK1 and IKK,was recently found to reduce cell proliferation,migration,and invasion by decreasing autophagy and the inhibition of melanoma-relevant proteins (Möller et al.,2020).An earlier clinical trial also demonstrated the glucose-controlling effect of amlexanox in patients (Oral et al.,2017).Eskiocak et al.(2017) found a TBK1/IKK inhibitor in the field of melanoma.In addition,several nonspecific inhibitors have also been studied in the xenograft environment.Research has shown that BX795 inhibits the growth of oral squamous cell xenograft (Bai et al.,2015).Another study showed that a dual TBK1/IKK inhibitor (compound 1) could enhance anti-programmed cell death 1 ligan 1 therapy in a xenograft model (Jenkins et al.,2018).Based on these discoveries in cancer and metabolic diseases,we should further investigate the potential neuroprotective and antiinflammatory effects of TBK1/IKK inhibitors,particularly in NTG.Moreover,a novel technology termed proteolysis-targeting chimera (PROTAC) has become increasingly popular in recent years;this technology can be used to target a specific protein for degradation.Indeed,PROTAC has been utilized for the selective degradation of TBK1 (Crew et al.,2017).Thomson et al.(2019)discovered a highly selective TBK1 inhibitor,termed GSK8612.This small molecule drug can be used to intercept the effect of TBK1 in the process of immunity,neuroinflammation,obesity,and cancer.Therefore,we investigated the functional roles of a TBK1 PROTAC and its therapeutic potential for NTG(Figure 1
).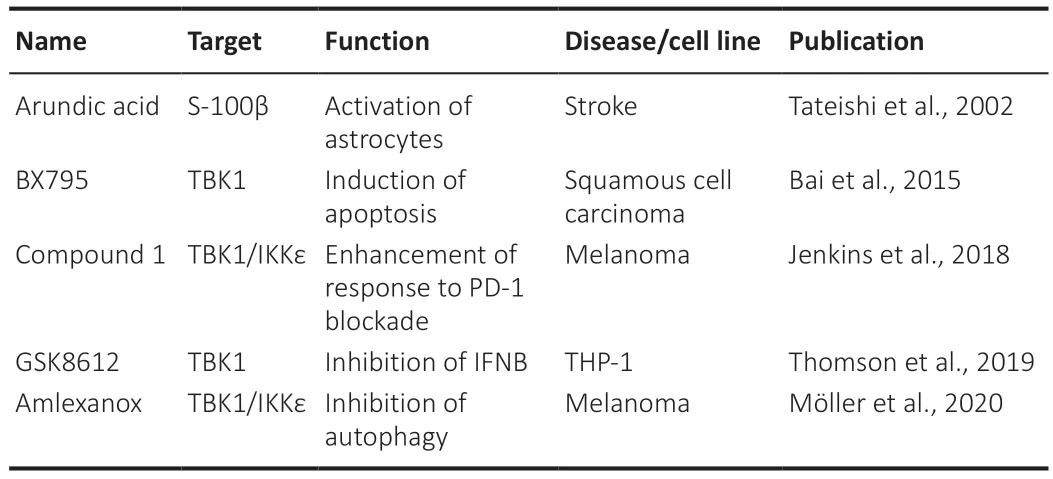
Table 1 | Summary of small molecule agents for the treatment of non-glaucoma disease
Glutamate Toxicity
Molecular mechanism underlying glutamate toxicity
Glutamate plays an important role in GON.It is thought that glutamate and the related excitatory amino acids (EAAs) activate glutamate receptors,which is followed by mediation of excitatory synaptic transmission at photoreceptor/bipolar cell synapses and bipolar/ganglion cell synapses (Sucher et al.,1998).In RGCs,glutamate neurotoxicity is induced by the over-stimulation of N-methyl-D-aspartate (NMDA) membrane receptors by glutamate.In this process,intracellular Cainflux is enhanced,which can cause organelle and DNA damage,including mitochondria and endoplasmic reticulum (Munemasa and Kitaoka,2013).Glutamate transporter was previously regarded as the only mechanism for eliminating glutamate from the extracellular fluid of the retina(Danbolt,2001).There are three transporters,namely glutamate transporter 1 (GLT-1),excitatory amino acid carrier 1 (EAAC1),and glutamate/aspartate transporter (GLAST) (Rauen,2000).Notably,EAAC1-and GLAST-KO mice have shown progressive RGC loss and GON without elevation of IOP (Harada et al.,2007).To explore the relationship between mutation in the GLAST gene and susceptibility to glaucoma,Yanagisawa et al.(2020) sequenced the EAAT1 gene of patients with glaucoma.They identified four heterozygous mutations(A169G,E219D,T318A,and A329T),which caused amino acid substitutions in the EAAT1 protein.Furthermore,A169G and A329T mutations impaired glutamate uptake;however,E219D and T318A mutations did not have an effect on EAA1.Moreover,GLAST-KO mice have been widely used to elucidate the mechanism underlying the development of NTG and provide potential therapeutic targets (Kimura et al.,2015;Dong et al.,2016;Sano et al.,2019).Similarly,EAAC1-KO mice are suitable animal models for studying NTG with the characteristics of sporadic and age-dependent pathology (Harada et al.,2007).An earlier study has shown that apoptosis signal-regulating kinase 1(ASK1) plays an important role in stress-induced apoptosis of RGCs in GLASTKO mice,which can be beneficial in the treatment of NTG (Harada et al.,2010).Therefore,EAAT1/GLAST is responsible for the pathogenesis of NTG and a potential therapeutic target (Harada et al.,2020).
Potential therapeutic agents
As mentioned above,the over-stimulation of NMDA receptors led to neuronal cell death in the inner retina through the process of retinal excitotoxicity(Parsons et al.,1998).Using the NMDA-induced retinal toxicity model,several studies have evaluated the effect of clinical drugs (Fang et al.,2010).Lal and Forster (1990) demonstrated that β-estradiol exerts a protective effect against NMDA-induced neurotoxicity.This finding suggests the potential effect of β-estradiol in the treatment of NMDA-related diseases,such as retinal ischemia and glaucoma.Another study utilizing the NMDA-induced retinal neurotoxicity model has shown a neuroprotective effect following the attenuation of delta9-tetrahydrocannabinol and cannabidiol,which is mediated by peroxynirite (El-Remessy et al.,2003).It has been demonstrated that NMDA antagonists are effective in preventing neuronal degeneration in Alzheimer’s disease (Farlow,2004).Researchers also focused on the effect of NMDA antagonists on RGCs (Takeda et al.,2018;Jiang et al.,2019).In Brown Norway rats,RGC degeneration,upstream changes in the optic nerve actin cytoskeleton,and the associated deterioration in visual function mediated by NMDA receptors can be reversed by treatment with MK-801 (a NMDA receptor blocker) (Omodaka et al.,2014).
Owing to the characteristics of GLAST-deficient mice,drugs that are capable of increasing the levels of GLAST may be useful for neuroprotection.Arundic acid has some beneficial effects that are associated with the suppression of delayed extracellular glutamate accumulation in the cerebral artery occlusion model (Tateishi et al.,2002).Additionally,it has been demonstrated that adenosine receptor A2AR antagonists enhance the recovery of retinal function following an ischemic attack (Zhong et al.,2013).A recent study has shown the activating effect of SCH442416 (an A2AR antagonist),which increased glutamate uptake in Müller cells (Li et al.,2015).Furthermore,cyanin chloride (a type of anthocyanin) prevents hyperbaric pressure-induced death of Müller cells by increasing the levels of GLAST (Chen et al.,2018b).Together,the above findings suggest that some clinical drugs and receptorrelated antagonists may be useful in the management of glaucoma.This evidence allows us to further investigate potential treatments for NTG (Figure
1
,Tables 1
and2
).
Table 2 | Summary of small molecule agents for the treatment of glaucoma
Stress
Molecular mechanism of oxidative stress in NTG
Oxidative stress refers to an imbalance between the production of reactive oxygen species (ROS) and antioxidant defenses,during which oxidative processes are superior to antioxidant systems (Kimura et al.,2017).It has been recognized as one of the pathogenic factors of glaucoma.Notably,the plasma levels of glutathione (GSH) are decreased in primary open-angle glaucoma,including NTG (Park and Moon,2012;Goyal et al.,2014).Oxidative stress is also involved in NTG-related GON (Trivli et al.,2019).The reduction in oxygen concentration leads to ROS formation,increasing the concentration of hydrogen peroxide (HO).This process further impairs electron flow and induces mitochondrial dysfunction.ROS formation induces oxidative damage to the mitochondria,and impairs cellular proteins and the DNA of RGCs as well as their axons (Chrysostomou et al.,2013).Additionally,the impairment of antioxidant systems,leads to over-production of ROS and mitochondrial dysfunction in lamina cribrosa cells (McElnea et al.,2011).Moreover,a recent study revealed that oxidative stress was increased and blood flow was decreased in glaucomatous marmosets;of note,both the retinal expression and blood repression of oxidative stress marker 4-hydroxynonenal were higher compared with those recorded in the control group (Noro et al.,2019).
Inhibition of oxidative stress and antioxidants
An increasing number of studies in animal models demonstrate that antioxidants are potential candidates for glaucoma therapy,supporting the hypothesis that suppression of oxidative stress increases RGC survival (Yang et al.,2016;Goichi et al.,2017).ASK1,which plays an essential process in oxidative stress-induced apoptosis,is activated by various oxidants (e.g.,HO)through the activation of the ASK-JUN N-terminal kinase/p38 (ASK-JNK/p38)pathway (Matsuzawa et al.,2002).ASK1 deletion may also reduce the level of factors that cause oxidative stress,such as tumor necrosis factor-alpha (TNF-α),which mediates neurodegeneration in glaucoma (Osaka et al.,2007;Guo et al.,2010).We need to further confirm the effect of ASK1 inhibitors on animal models of NTG.It has been shown that dedicator of cytokines 3 (Dock3;an atypical guanine exchange factor) increase RGC survivalin vivo
(Namekata,2004).Dock3 may prevent oxidative stress-induced RGC death by inhibition of the ASK1 pathway (Bessero and Clarke,2010).Valproic acid (a short-chain fatty acid) has been used clinically in the treatment of various conditions,exhibiting a relatively good safety profile (Namekata et al.,2013).It can reduce the oxidative levels in RGCs through activation of the brain derived neurotrophic factor-tropomysin related kinase B (BDNF-TrkB) pathway in GLAST-KO mice (Kimura et al.,2015).Studies on retinitis pigmentosa showed that oral administration of valproic acid improved visual function (Kimura et al.,2015;Iraha et al.,2016).Further studies are required to assess the efficacy and safety of valproic acid for the treatment of NTG.Similarly,it was shown that N-acetylcysteine (an antioxidant and precursor of GSH) protects RGCs in EAAC1-KO mice by increasing the levels of retinal GSH and reducing oxidative stress (Sano et al.,2019).Moreover,N-acetylcysteine has been associated with a relatively good safety profile,with mild side effects (Atkuri et al.,2007).Topical application of coenzyme Q10 (CoQ10) (an important antioxidant) also protected RGCs in a rat model.This evidence can help scholars to investigate the additional effects of CoQ10 as a hypotensive drug for patients with glaucoma (Davis et al.,2017;Quaranta et al.,2019).Moreover,a recent study involving a mouse model of NTG revealed the anti-oxidative stress effects of astaxanthin against RGC degeneration (Kikuchi et al.,2020).Collectively,these discoveries in animal models provide us with novel candidates for the treatment of NTG in the future (Figure 1
andTable 2
).Several clinical studies have investigated the systemic oxidative damage and antioxidant status in patients with NTG.Yilmaz et al.(2016) demonstrated that hyperlipidemia,oxidative stress,and variations in phenotype distribution of paraoxonase 1 may play important roles in the pathogenesis of NTG.These results suggested that hyperlipidemia is associated with NTG.Increased levels of serum total antioxidant and decreased concentration of 8-hydroxy-2’-deoxyguanosine (a marker for systemic mutagenizing DNA lesions) were found in patients with NTG,reflecting compensatory alternations in response to an increased oxidative stress state (Yuki et al.,2010).Moreover,it has been suggested that dietary antioxidants are effective in decelerating the progression of glaucoma (Mozaffarieh et al.,2008).A case study reported that the combination of once-daily dietary supplementation (containing a citicoline,homotaurine,and vitamin E) with topical medications improved the visual field in a patient with NTG (Verdina et al.,2020).Interestingly,some experimental studies on glaucoma provided evidence regarding the effects of citicoline on RGCs without altering the IOP (Faiq et al.,2019).A recent report also demonstrated the role of citicoline in protecting neural tissues and visual function in glaucoma beyond IOP control (van der Merwe et al.,2021).The therapeutic role of niacin,also termed vitamin B3,was supported by studies examining patients with NTG (Jung et al.,2018).Baillargeon and Sonnet(2010) identified the physiological and molecular mechanisms underlying the effectiveness of Ginkgo biloba extract in NTG therapy.Polyphenolic flavonoids– the molecules contained in Ginkgo biloba extract– prevent oxidative stress in mitochondria and protect RGCs from apoptosis (Ghiso et al.,2013).Based on its antioxidant activity and other beneficial effects (e.g.,increased ocular blood flow,inhibition of platelet-activating factor,and neuroprotection),Ginkgo biloba extract may be valuable in the treatment of NTG (Saccà et al.,2019).
Neuroinflammation and Immunity
Immune system in glaucomatous neurodegeneration
The eye is regarded as an immune privileged site,which can be affected by diseased conditions (e.g.,glaucoma).In this process,the blood-retina barrier is destroyed and cytokine production is altered (Perez and Caspi,2015).Several studies have demonstrated that numerous factors (e.g.,oxidative stress,vascular endothelial growth factor,and inflammation) can affect the permeability of the blood-retina barrier (Kaur et al.,2008;Kaur and Ling,2008).In glaucoma,inflammation may be induced by an altered crosstalk between RGCs and neuroglia cells,involving the release of pro-inflammatory mediators,such as ROS,nitric oxide,TNF-α,and interleukin-1β (IL-1β)(Chua et al.,2012;Masuda et al.,2017).As the first line of defense against pathogens,the innate immunity offers a rapid yet short response to infection.In glaucoma,the elevated IOP can trigger an innate immune response (Wei et al.,2019).Using an experimental model of glaucoma,several researchers observed increased microglia activity,cell density,and expression of the complement of C1q in the retina and optic nerve,particularly prior to RGC and axonal loss (Howell et al.,2011;Trost et al.,2021).In contrast to the innate immune system,the adaptive immune responses involve T and B lymphocytes,require up to 7 days for activation,and are also featured in glaucomatous pathogenesis (Jiang et al.,2020).Following the elevation of IOP,heat shock proteins (HSPs) are upregulated and significantly increased in human glaucomatous retinas (Soto and Howell,2014).Chen et al.(2018a)reported that HSP-specific memory T cells are induced by commensal microflora,and activated by host HSPs released in the retina,offering key evidence for the involvement of autoimmunity in glaucoma (Chen et al.,2018a).Moreover,elevated numbers of CD3CD8lymphocytes were observed in both NTG and primary open-angle glaucoma.Particularly,CD8HLA-DRlymphocytes were more prevalent in NTG.Further studies on the immunity response in NTG are warranted to elucidate the mechanism of neurodegeneration.
Microglial cells (immune cells residing in neural tissue) are responsible for synaptic maintenance and immune response to injury and inflammation(Colonna and Butovsky,2017;Salter and Stevens,2017).These cells act as neuropathological sensors and a first line of defense to injury in the CNS,including the brain and retina (Wei et al.,2019).Both adenosine 5’triphosphate (ATP) release and microglial activation induce the mechanical strain accompanying the elevation of IOP (Campagno et al.,2021).As immunomodulatory cells,microglial cells trigger an immune response in glaucoma,playing a neuroprotective or neurotoxic role through two phenotypes,namely M1 (pro-inflammatory) or M2 (neuroprotective)(Fernandez-Albarral et al.,2022).However,in ocular hypertension eyes,the majority of microglial cells did not exhibit the M2 phenotype or a neuroprotective effect (de Hoz et al.,2013).
Research progress in neuroprotection
NTG has been linked to several conditions,including nocturnal hypotension,inflammatory diseases,and alterations in C-reactive protein.Atalay et al.(2019) evaluated the neutrophil-to-lymphocyte and platelet-to-lymphocyte ratios,C-reactive protein levels,and erythrocyte sedimentation rate in NTG.There were no significant differences in neutrophil-to-lymphocyte and platelet-to-lymphocyte ratios found,providing insight into the differential diagnosis of NTG.More interestingly,several researchers have discovered novel small molecule agents,which may control neuroinflammation and protect the optic nerve.Recent research has revealed that SCH 58261 (a potent and selective antagonist for human A2AR) prevents morphological alterations and ROS production in microglial cells,proving its role in controlling retinal neuroinflammation (Ongini et al.,1999;Aires et al.,2019).Moreover,in an inducible mouse model of glaucoma,a small peptide inhibitor of the Fas receptor (ONL1204) provided robust neuroprotection by abrogating the activation of microglial cells and inhibiting the induction of multiple cytokines and chemokines,components of the complement cascade,toll-like receptor pathway,and inflammasome pathway (Krishnan et al.,2019).Furthermore,some studies focused on the mechanisms regulating the activation of microglial cells,and attempted to identify potential targets for neuroprotective therapy.Wang et al.(2021) reported that the miR-93/signal transducer and activator of transcription 3 (miR-93/STAT3) pathway is directly related to the downregulation of retinal microglia-mediated neuroinflammation.Matrix-bound nanovesicles are a distinct class of extracellular nanovesicles localized specifically to the extracellular matrix.Studies have verified that these nanovesicles preserve visual function in a rat model (van der Merwe et al.,2019).Activation of the complement cascade has been observed in patients with glaucoma and models of glaucoma(Silverman et al.,2016;Bosco et al.,2018;Reinehr et al.,2018).Therefore,the use of therapies targeting the inflammation-related and specific complement pathways may improve the effectiveness of neuroprotective strategies (Figure
1
andTable 2
).ET-1 and Vasoconstriction
Microcirculation is mainly regulated by vasoregulatory factors from endothelial cells,including nitric oxide and ET-1 (Haefliger et al.,2001).ET-1 is a potent 21-amino acid vasoconstrictive peptide produced by vascular endothelial cells (Inoue et al.,1989).The vasoconstrictive effect of ET-1 is mainly mediated by ET-1-specific ETA receptors,which are located on vascular smooth muscle cells (Sakurai et al.,1990).The association between plasma ET-1 levels and the pathogenesis of NTG has been previously reported (Cellini et al.,1997).It has been proposed that higher ET-1 levels may be related to the primary stage of visual field loss (Sugiyama et al.,1995).A recent study showed that age has a significantly greater influence on endothelin plasma levels in patients with NTGversus
those with high tension glaucoma (Konieczka et al.,2020).Collectively,the above evidence supports that endothelin plays vital roles in the pathogenesis of glaucoma,particularly in NTG (Figure
1
).In NTG,Flammer syndrome is the most important cause of vascular dysregulation (Konieczka et al.,2014).Endothelial dysfunction impairs ocular blood flow and promotes vasospasm and GON,as a result of NTG (Trivli et al.,2019).In addition,researchers have investigated the impact of endothelinrelated genetic polymorphisms on glaucoma.Polymorphic varieties of ET-1(K198N) and ET-1 receptor type A (C1222T) affect plasma concentration of endothelin and influence the IOP and arterial blood pressure in patients with NTG.Notably,there was no association found between the plasma levels of endothelin and risk factors for NTG (Wróbel-Dudzińska et al.,2015;Kosior-Jarecka et al.,2016a).Nevertheless,these studies are limited because they involved only one population or were conducted in one geographic region.Further studies are warranted to assess the functional impact of these genetic polymorphisms in a higher number of patients.Recently,more studies have concentrated on further mechanisms underlying the regulation of RGC death by ET-1.As previously shown,JNK signaling mediates neurodegeneration in glaucoma (Syc-Mazurek et al.,2017;Ye and Meng,2021).Marola et al.(2020) indicated that ET-1 can induce RGC death through the JUN-dependent pathway.ET-1 also activates c-Jun,and the DNA binding of c-Jun and CCAAT/enhancer-binding protein β (C/EBPβ) further indicates that ET-1 triggers c-Jun through endothelin receptors and c-Jun feedback elevates the expression of endothelin receptor (Wang et al.,2017).Further investigation is warranted to determine the upstream regulators and downstream targets of JUN signaling.This evidence may help to elucidate the mechanism underlying the regulation of RGC death by endothelin.
Conclusions
In this review,we discuss and summarize the results of recent research on various mechanisms involved in the regulation of RGC death in NTG,and strategies for the inhibition of RGC death and protection of the optic nerve(Figure 2
).Several small molecule agents related to autophagy,glutamate neurotoxicity,and neuroinflammation,as well as some clinical antioxidants inhibiting oxidative stress,have been identified.ET-1 and related signaling pathways are also analyzed to reveal novel therapeutic candidates for NTG.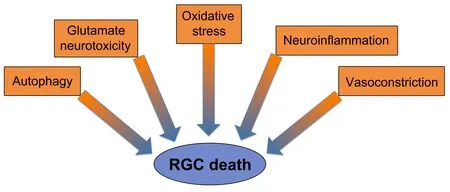
Figure 2 | Risk factors contributing to RGC death in glaucoma.
The pathogenesis of NTG involves multiple factors.NTG is a type of glaucoma associated with progressive optic neuropathy in the absence of elevated IOP.Hence,therapeutic efforts should be focused on alleviating RGC death and protecting the optic nerve.The mechanisms involved in the regulation of RGC death are attracting considerable research attention.It has been demonstrated that several small molecule agents and clinical antioxidants are effective in animal models or patients.Additional studies on these small molecules with neuroprotective properties are required for the discovery of potential therapeutic targets in patients with NTG.
Limitations and Perspectives
The limitations of the present review and several novel progresses in vision restoration should be acknowledged.Stark and Caprioli (2016) and Gu et al.(2021) focused on axon regeneration and reported that visual acuity may be reversed.A recent study showed that reticulon 3 (RTN3) can enhance neuroprotection and CNS axon regeneration,particularly RGC axon regeneration,thus improving visual function (Alhajlah et al.,2021).In addition,Pfeiffer et al.(2019) described their early findings regarding retinal remodeling retinal pathoconnectome 1 (RPC1),which provides the perspective of computational molecular phenotyping in retinal reconstruction(Pfeiffer et al.,2019).Further studies focusing on structural neuroprotection are warranted.Such studies may assist in developing earlier and more effective strategies for reversing neural damage in patients with NTG.
Author contributions:
Manuscript writing:WCS;manuscript design and review,and data collection:BQH,JY.All authors approved the final manuscript.
Conflicts of interest:
The authors declare no conflicts of interest.
Availability of data and materials:
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open access statement:
This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:
Sanjoy K.Bhattacharya,University of Miami School of Medicine,USA.
Additional file:
Open peer review report 1.

- 中国神经再生研究(英文版)的其它文章
- Neuroaxonal and cellular damage/protection by prostanoid receptor ligands,fatty acid derivatives and associated enzyme inhibitors
- Extracellular vesicles in Alzheimer’s disease:from pathology to therapeutic approaches
- Molecular approaches for spinal cord injury treatment
- Sex-biased autophagy as a potential mechanism mediating sex differences in ischemic stroke outcome
- Adipose tissue,systematic inflammation,and neurodegenerative diseases
- Interleukin-1:an important target for perinatal neuroprotection?