
The regulatory role of Pin1 in neuronal death
2022-05-25 09:26ShuChaoWangXiMinHuKunXiong
中国神经再生研究(英文版) 2023年1期
Shu-Chao Wang,Xi-Min Hu,Kun Xiong,
Abstract Regulated cell death predominantly involves apoptosis,autophagy,and regulated necrosis.It is vital that we understand how key regulatory signals can control the process of cell death.Pin1 is a cis-trans isomerase that catalyzes the isomerization of phosphorylated serine or threonine-proline motifs of a protein,thereby acting as a crucial molecular switch and regulating the protein functionality and the signaling pathways involved.However,we know very little about how Pin1-associated pathways might play a role in regulated cell death.In this paper,we review the role of Pin1 in regulated cell death and related research progress and summarize Pin1-related pathways in regulated cell death.Aside from the involvement of Pin1 in the apoptosis that accompanies neurodegenerative diseases,accumulating evidence suggests that Pin1 also plays a role in regulated necrosis and autophagy,thereby exhibiting distinct effects,including both neurotoxic and neuroprotective effects.Gaining an enhanced understanding of Pin1 in neuronal death may provide us with new options for the development of therapeutic target for neurodegenerative disorders.
Key Words:apoptosis;autophagy;calpain;central nervous system;necroptosis;necrosis;neurodegenerative diseases;neuron;Pin1;regulated neuronal death
Introduction
Cell death is a fundamental process that is essential for the normal development and homeostasis of organisms (Bai et al.,2020;Moujalled et al.,2021).In general,there are three types of cell death:apoptosis,autophagy,and necrosis (Carloni et al.,2017;Bai et al.,2020).Apoptosis refers to regulated cell death (RCD) that occurs in developing tissues and plays an important role in homeostasis.RCD is characterized by chromatin condensation,nuclear fragmentation,and the formation of apoptotic bodies.Previous studies have reported that a large number of apoptotic proteins play a role in apoptotic signaling pathways,including caspases and B-cell lymphoma 2 (Bcl-2) family members (Bai et al.,2020;Özel et al.,2021).Autophagy is another form of RCD during which cellular contents,such as organelles,are encapsulated by a double-membrane to form auto-phagosomes that are subsequently degraded by lysosomes (Napoletano et al.,2019;Bourdenx et al.,2021).Autophagy plays an important role in maintaining homeostasis and protecting cells against stressful stimuli.Traditionally,necrosis is considered to represent a form of accidental cell death that is characterized by membrane destruction,the release of internal materials,and inflammation (Chen et al.,2021;Hu et al.,2021b).However,recent research has provided evidence that some types of necrosis also can be regulated at the molecular level (Theobald et al.,2021;Tonnus et al.,2021).Other researchers have reported that this form of regulated necrosis (RN) includes necroptosis (Evans and Coyne,2019;Yuan et al.,2019),pyroptosis (Lammert et al.,2020),and neutrophil cell death(Sung and Hsieh,2021;Yan et al.,2021).RCD,featuring apoptosis,autophagy and RN,is recognized to be beneficial to the host by eliminating certain intracellular pathogens or stimuli (Guo et al.,2020;Wang et al.,2020b;Yan et al.,2021).It is vital that we know how RCD responds to pathogens or injuries.
Prolylcis
-trans
isomerase NIMA-interacting 1 (Pin1) belongs to the parvulin subfamily and is an 18-kDa protein with a C-terminal PPIase domain and a N-terminal WW domain (Kumari et al.,2021;Tonnus et al.,2021;Zheng et al.,2021).Pin1 is able to bind and then isomerize phosphorylated serine-proline or phosphorylated threonine-proline (pSer/Thr-Pro) motifs,thus regulating a series of biological processes (Koikawa et al.,2021;Kumari et al.,2021).The WW domain responds to and binds to pSer/Thr-Pro sequences,while the catalytic PPIase domain isomerizes the prolyl bond in the pSer/Thr-Pro motif;collectively,these mechanisms play an important role in many biological processes,such as the cell cycle,immune responses,and tumorigenesis(Koikawa et al.,2021;Kumari et al.,2021).In addition to binding to the prolyl bond in the pSer/Thr-Pro motif,Pin1 can also specifically regulate the activity of mitotic and nuclear proteins in a phosphorylation-dependent manner(Kumari et al.,2021).Pin1 is predominantly expressed in cell nuclei and functions as a mitotic regulator,thus playing a vital role in DNA replication and mitosis (Zhang and Zhang,2019;Kumari et al.,2021).The targets of Pin1 include not only proteins that exert function in transcription and the cell cycle,but also those involved in many physiological and pathological processes,including apoptosis,proliferation,and maintenance of the cytoskeleton (Thorpe et al.,2004;Lepore et al.,2021).Pin1 acts as a context-specific signal transducer based on specific environmental signals (Oster et al.,2021).Researchers have found that Pin1 plays a dual role in cellular apoptosis (Baik et al.,2015;Dubiella et al.,2021;Fagiani et al.,2021).For example,Pin1 was shown to enhance the anti-cell death ability of Bcl-2 and inhibit apoptosis by directly inactivating Bcl-2 associated X (Bax) (Bianchi et al.,2019;Makinwa et al.,2020).However,Pin1 has also been shown to act as a promoter of cell apoptosis by enhancing the expression of pro-apoptotic genes,such asp53
(Balaganapathy et al.,2018;Hleihel et al.,2021).Pin1 has been reported to be distributed in the central nervous system (CNS) and participate in a variety of neurodegenerative diseases,including Alzheimer’s disease (AD),Parkinson’s disease (PD),amyotrophic lateral sclerosis (ALS) and Huntington’s disease (HD) (Ghosh et al.,2013;Carnemolla et al.,2017;Iridoy et al.,2018;Napoletano et al.,2021;Samimi et al.,2021).An increasing body of evidence now supports the fact that Pin1 could exert a neuroprotective role or act as a promoter for neuronal death by upregulating survival-promoting factors or by downregulating death-suppression factors.However,the comprehensive regulatory pathways underlying the specific role of Pin1 in RCD has yet to be elucidated.In this review,we set out to summarize our current knowledge of Pin1 in neuronal RCD and discuss the critical regulatory mechanisms associated with Pin1 in the CNS (Additional Table 1
).In addition,Pin1 is an intriguing target for cancer therapy at present and could represent a desirable pharmaceutical target for neurological therapy.As such,we also summarize research relating to Pin1-specific inhibitors.Search Strategy
We searched all literatures published from June 1998 to May 2021 that relates to the role of Pin1 in neuronal death (Figure 1
) in this narrative review.We performed a PubMed search using the term“Pin1”AND“neuron”to identify potential neuronal death in neurodegenerative diseases that are mediated by Pin1 dysfunction.We next screened the results by the title,keywords,abstract and excluded non-neuronal death-related articles,and retrieved further papers by citation tracking.Apoptosis
Apoptosis is controlled by several apoptotic genes and characterized by RCD(Bai et al.,2020;Hu et al.,2021a;Nascimento et al.,2022).Traditionally,apoptosis can be divided into two pathways,the extrinsic and intrinsic pathways (Carneiro and El-Deiry,2020;Nascimento et al.,2022).In the extrinsic pathway,apoptosis is induced by extracellular signals which then trigger the activation of caspases and caspase-mediated pathways (Carneiro and El-Deiry,2020;Nascimento et al.,2022).The intrinsic pathway is activated by intraocular stimulation,such as signals from the mitochondria;this leads to the activation of caspases and eventually causes the degradation of vital proteins,thus inducing apoptosis (Carneiro and El-Deiry,2020;Nascimento et al.,2022).The role of Pin1 in neuronal apoptosis has been investigated in many studies (Figure 2
) (Becker and Bonni,2007;Moujalled et al.,2021).For example,Pin1 has been reported to mediate neuronal apoptosis by promoting the expression of pro-apoptotic genes,such as p53 (Grison et al.,2011;Balaganapathy et al.,2018).Literature indicates that Pin1 may participate in neuronal survival or apoptosis,depending on the specific development stages of neurons as well as the different tissues and diseases involved.In this regard,herein,we discuss the different roles of Pin1 in apoptosis in different neurodegenerative diseases.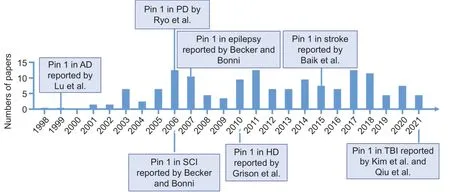
Figure 1 | Timeline of the milestones of Pin1-mediated neuronal death in neurodegenerative diseases.
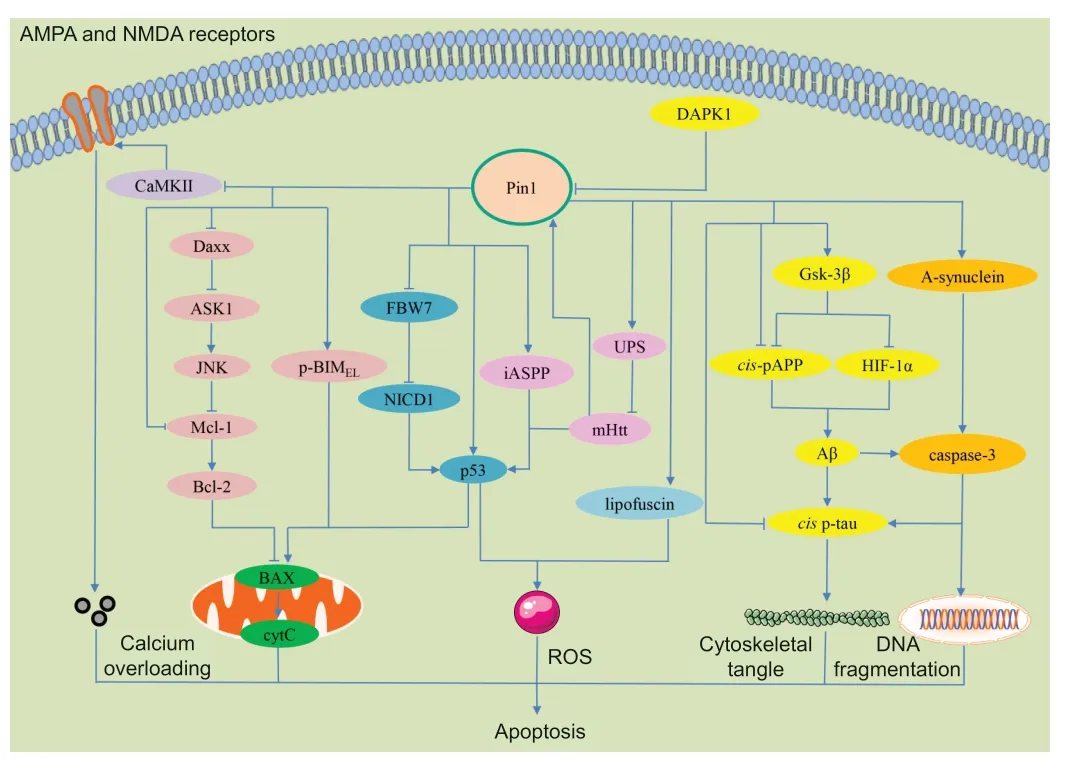
Figure 2 | The molecular mechanisms underlying Pin1-regulated apoptosis signaling pathways.
Pin1 mediates neuronal apoptosis in AD
AD is a leading dementia disease and affects the global population.AD is characterized by the accumulation of β-amyloid peptide (Aβ) with hyperphosphorylated tau (p-Tau),a microtubule associated protein (Azimi et al.,2017;Gu and Liu,2020;Li et al.,2021b;Blanchard et al.,2022).Lu et al.(1999) were the first to report that Pin1 was involved in the process of AD.In the normal human brain,Pin1 is mainly distributed in the neuronal nuclei.However,in AD patients,Pin1 is localized in both the neuronal cytoplasm and perikaryal neurofibrillary tangles (NFTs);furthermore,the levels of Pin1 are significantly reduced (by 39%) in the brains of AD patients when compared to normal brains (Pastorino et al.,2006;Samimi et al.,2021).Analysis of Pin1 knockout mice showed that the accumulation of p-Tau,neuronal loss,and behavioral deficits appeared together (Thorpe et al.,2004;Azimi et al.,2017).Other research has shown that Pin1 is able to bind to the phosphorylated Thr231 motif of Tau (pT231-Tau) and can accumulate in NFTs within the neuronal cytoplasm (Lu et al.,1999).In addition,Pin1 can ameliorate p-Tau pathology by isomerizing p-Tau,thus recovering its ability to bind to and restore cytoskeletal integrity (Hashemzadeh-Bonehi et al.,2006;Kesavapany et al.,2007).It has been reported that p-Tau exists in two isoforms,physiologicaltrans
and pathologicalcis
(Wang et al.,2020a;Qiu et al.,2021).The trans form of p-Tau exhibits normal functionality and may undergo degradation.In contrast,thecis
form of p-Tau is abnormal and cannot be degraded;this form has a high tendency for aggregation (Zheng et al.,2021).Pin1 can accelerate the transformation of thecis
isoform to thetrans
isoform of pT231-Tau and restore its normal function.In fact,cis
pT231-Tau is also significantly increased in human AD brains and is strongly correlated with NFTs and reduced levels of Pin1 (Nakhjiri et al.,2020;Wang et al.,2020a;Qiu et al.,2021).A large body of evidence now suggests that interaction between Aβ and Tau could enhance the process of AD (Selkoe and Hardy,2016;Shentu et al.,2018).For example,Pastorino et al.(2006) reported that Pin1 could modulate the production of Aβ production.Enhanced Aβ production can also promote the phosphorylation of Tau and the generation of NFTs (Dakson et al.,2011).In particular,Pin1 binds specifically to the phosphorylated Thr668-Pro motif of amyloid precursor protein (APP) (pT668-APP) and catalyzes pT668-APP fromcis
to trans transformation (Nowotny et al.,2007;Dakson et al.,2011).Thecis
pT668-APP isoform promotes the processing of Aβ.By catalyzing from acis
totrans
isoform,Pin1 could inhibit the processing of Aβ (Nowotny et al.,2007;Dakson et al.,2011).The deletion of Pin1 in mice has been linked to the accumulation of Aβ and neurotoxicity (Wang et al.,2020a;Li et al.,2021a).Some other reports have also indicated that molecules leading to Tau phosphorylation,including caspase-3,can also be activated by Aβ accumulation,finally resulting in neuronal death and the progression of AD process (O’Brien and Wong,2011;Rong et al.,2017).Glycogen synthase kinase-3β (GSK-3β),a serine/threonine protein kinase,plays a role in several vital cellular functions,such as growth and glycogen metabolism and can also promote hypoxia-inducible transcription factor-1 degradation (Flügel et al.,2012).Recently,Lonati et al.(2014) reported that in cortical neurons,hypoxia-inducible transcription factor-1 was maintained at higher levels in the brains of AD patients;this was coincident with reduced levels of GSK-3β protein.Interestingly,these authors found that reduced levels of Pin1 inhibited GSK-3β enzymatic activity,ultimately inhibiting hypoxia-inducible transcription factor-1 protein degradation and contributing to Aβ production and progression in AD patients (Lonati et al.,2014).As the levels of Pin1 are decreased in AD patients and because Pin1 plays a vital role in AD processing in animal models of AD,it follows that the down-regulation of Pin1 might be an early potential biomarker for the progression of AD (Xu et al.,2017).Pin1 mediates neuronal apoptosis in TBI
TBI includes closed-head injuries (in which the skull and dura are intact),penetrating injuries (in which skull and dura are broken),and injury to the brain parenchyma (Wood,2018).TBI is characterized by acute neuronal dysfunction and is thought to be a non-aging risk factor for AD (Wood,2018).Tau pathology is also a pathophysiological factor in TBI;patients with TBI have higher levels of tau (Shin et al.,2021).Furthermore,recent studies have shown that in the brains of TBI patients,Pin1 is widely distributed in the neurons and correlates with neurodegenerative pathologies (Kim et al.,2021;Qiu et al.,2021).Other research has shown thatcis
pT231-Tau,but not thetrans
form,contributes to neuronal pathology and functional impairment in a model of TBI (Albayram et al.,2017,2018;Qiu et al.,2021;Tonnus et al.,2021).Recent studies showed that death-associated protein kinase 1 (DAPK1)plays a critical role in regulating cis pT231-Tau after TBI (Kim et al.,2021;Qiu et al.,2021).DAPK1 is a calcium/calmodulin-dependent serine/threonine(Ser/Thr) kinase that plays a critical role in neuronal death.DAPK1 has been shown to increase significantly in the brains of a mouse model of TBI and then promotes the induction ofcis
pT231-Tau (Kim et al.,2021;Qiu et al.,2021).Other research has shown that DAPK1 is a key regulator of the activity of Pin1.DAPK1-mediatedcis
pT231-Tau transformation has been proven to be regulated by Pin1 in the brain after injury (Kim et al.,2021;Qiu et al.,2021).Following brain damage,the activity of Pin1 is known to decrease;this is accompanied by an increase in the expression ofcis
pT231-Tau,thus promoting neuronal death (Kim et al.,2021;Qiu et al.,2021).Furthermore,the pharmacological inhibition of DAPK1 activity significantly increases the levels of Pin1 and significantly decreases the expression ofcis
pT231-Tau and the extent of neuronal injury (Kim et al.,2021;Qiu et al.,2021).Thus,DAPK1-Pin1-cis
pT231-Tau is a novel regulatory pathway in TBI,and exerts an important effect on the progression of TBI.Thus,the targeting of this pathway might be a potential therapeutic method for TBI (Tonnus et al.,2021).It should be noted,however,that the levels of Tau change in both AD and TBI(Blennow et al.,2016).In AD,Aβ aggregation induces tau phosphorylation and accumulation.However,in TBI,Aβ has no effect on TBI-related tau accumulation.Thus,the mechanisms of tau accumulation in TBI might be distinct from AD (Blennow et al.,2016).Pin1 mediates neuronal apoptosis in PD
PD is a disease that is characterized by the loss of dopaminergic neurons and the formation of α-synuclein aggregates in Lewy bodies.Pin1 also has been reported to be implicated in the pathogenesis of PD (Matena et al.,2013;Ibáñez et al.,2014).Ryo et al.(2006) were the first to report that Pin1 is located in the Lewy bodies of PD patients.In particular,the overexpression of Pin1 may facilitate the formation and stability of α-synuclein aggregation in experimental models of PD (Ryo et al.,2006).Other research has shown that Pin1 is highly up-regulated in dopaminergic cells and nigrostriatal cells after Parkinsonian neurotoxicant 1-methyl-4-phenylpyridinium treatment (Ghosh et al.,2013).Furthermore,Pin1 knockdown or functional inhibition almost completely prevented 1-methyl-4-phenylpyridinium-induced caspase-3 activation,DNA fragmentation and neuronal death,thus indicating that Pin1 has a pro-apoptotic role and causes α-synuclein aggregation (Ghosh et al.,2013).However,until now,the precise regulatory mechanism underlying the upregulation of Pin1 in PD has yet to be fully elucidated.Ghosh et al.(2013)reported that the upregulation of Pin1 may be directly due to neurotoxic stress and may then contribute to the death of dopaminergic neurons.Therefore,the overexpression of Pin may mediate apoptosis and may represent a critical neurotoxic event in the pathogenesis of PD.
Pin1 mediates neuronal apoptosis in HD
HD is an autosomal dominant neurodegenerative disease caused by an expanded CAG repeat in the gene encoding huntingtin (Htt) protein;this condition is characterized by the death of medium spiny neurons in the striatum (Tabrizi et al.,2020).It has been reported that mutatedHtt
binds top53
,increases the levels of p53 protein,enhances the phosphorylation of p53 at Ser46 and evokes DNA damage in the striatum and cerebral cortex of patients with HD (Grison et al.,2011).It is important to note that the functionality of Pin1 plays a key role in stimulating the pro-apoptotic role ofp53
(Agostoni et al.,2016).First,mutated Htt promotes the activation of Pin1.Subsequently,Pin1 interacts withp53
and triggers dissociation from its inhibitor (inhibitor of apoptosis-stimulating protein ofp53
;iASPP),finally promoting the expression of a series of apoptotic genes (Grison et al.,2011).However,in contrast,another study found that Pin1 might act as a negative regulator for Htt accumulation in a genetic mouse model of HD (Carnemolla et al.,2017).These authors found that the overexpression of Pin1 led to a reduction ofHtt
accumulation and that this might occur via degradation by the ubiquitin-proteasome system (Carnemolla et al.,2017).Therefore,the role of Pin1 in neuronal apoptosis might have a different effect in that it may stimulateHtt
clearance-mediated neuronal protection and promotep53
mediated-apoptosis (Grison et al.,2011).Furthermore,by using a genetic model of Pin1 deletion,researchers found that the role of Pin1 in neuronal death might be time-dependent (Agostoni et al.,2016).In the early stages,Pin1 activation reduces the DNA damage response.However,in the middle stages of life,the activation of Pin1 might be triggered byHtt
,thus initiating the role of Pin1 inp53
-mediated neuronal apoptosis.In aging individuals,Pin1 significantly increases the amount ofHtt
accumulation (Agostoni et al.,2016).In summary,the role and mechanisms of Pin1 in neuronal apoptosis provides some support for the pathogenesis of HD.However,the role of Pin1 in HD still requires further research.Pin1 mediates neuronal apoptosis in stroke
Stroke is a disease involving sudden impairment of consciousness and motio;this condition is caused by the obstruction or rupture of blood vessels in the brain (Magnusson et al.,2014).Baik et al.(2015) reported that Pin1 contributes to the process of stroke through Notch signaling.The Notch signaling pathway plays a vital role in the development of the CNS,including neuronal differentiation,determination,and certain neural pathological diseases (Magnusson et al.,2014).Notch signaling has also been implicated in neuronal death (Arumugam et al.,2006;Magnusson et al.,2014).It has been reported that the levels of Pin1 and Notch intracellular domain 1 (NICD1),an active domain of Notch,were both increased following stroke (Baik et al.,2015).Further research showed that the overexpression of Pin1 could promote NICD1 stability and its pro-apoptotic function after stroke (Baik et al.,2015).It was also found that Pin1 binds to and stabilizes NICD1 domain through the WW domain (Baik et al.,2015).Following stroke,Pin1 interacts with NICD1 and enhances its stability by inhibiting F-box and WD repeat domain containing domain protein 7-mediated poly-ubiquitination,ultimately leading to NICD1-induced neuronal apoptosis (Wang et al.,2014;Baik et al.,2015).Other research indicated that Pin1 regulates p53 transactivation and the upregulation of pro-apoptotic genes,such asBax
,via the activation of NICD1,finally promoting neuronal apoptosis and stroke (Balaganapathy et al.,2018).The tumor suppressorp53
is an important decision maker in the cell cycle and apoptosis and responds to a series of stimulations.It has been reported that the phosphorylated form ofp53
induced by stress combines with the WW-domain of Pin1 to form a complex (Arumugam et al.,2018;Balaganapathy et al.,2018).Next,phosphorylatedp53
undergoes a conformational change that is catalyzed by Pin1,thus enhancing the transactivation activity ofp53
(Zacchi et al.,2002;Balaganapathy et al.,2018).The interaction between NICD1 and p53 is an important signaling step that induces neuronal apoptosis in stroke (Arumugam et al.,2018).The disturbance of NICD1/p53
interaction could ameliorate the process of stroke(Balaganapathy et al.,2018).In addition,some other reports indicated that the production of reactive oxygen species (ROS) was a critical effector in the Pin1-Notch-p53
pathway and plays a role in the activation of apoptosis in stroke (Li et al.,2019).Pin1 mediates neuronal apoptosis in SCI
SCI is a severe trauma that causes severe injury to the function of the motor and sensory neurons of the spinal cord and generally is associated with a poor prognosis (Yuan et al.,2021).Myeloid cell leukemia sequence-1 (Mcl-1),an anti-apoptotic factor of the Bcl-2 family,remains phosphorylated in the basal state (Carlet et al.,2021).In normal neurons,Pin1 binds with pT163-Mcl-1 and inhibits Mcl-1 ubiquitination and degradation (Li et al.,2017).However,after SCI injury,Mcl-1 undergoes degradation following the removal of Pin1,thus resulting in Bcl-2 inhibition and the release of cytochrome C into the cytosol(Li et al.,2007).Furthermore,the activation of c-Jun N-terminal kinase (JNK)induced by SCI can perturb the interaction between Pin1 and phosphorylated Mcl-1,thereby promoting the release of cytochrome C (Li et al.,2007).The JNK pathway plays a complicated role in many physiological and pathological processes,such as cell apoptosis and differentiation,and may be activated by various stress signals and inflammation (Zhang et al.,2021).JNK also can promote the mitochondrial apoptotic pathway by regulating the release of BAX under the control of Pin1 (Shen et al.,2009).Other reports have also indicated that Pin1 binds to the Ser178-Pro motif in phosphorylated death domain associated protein and promotes the degradation of death domain associated protein via ubiquitination (Ryo et al.,2007).Degraded death domain associated protein can interact with apoptosis signal-regulating kinase 1 and then activate the apoptosis signal-regulating kinase 1/JNK signaling pathway (Ryo et al.,2007;Oh and Mouradian,2018).Most importantly,Pin1 also binds with phosphorylated Bcl-2-interacting mediator of cell death,extralong (BIM) at Ser65 and JNK-interacting protein 3 to form a complex (Becker and Bonni,2006,2007).Pin1 also mediates the isomerization of BIMand leads to a conformational change in pSer65-BIM(Becker and Bonni,2006,2007).It has also been reported that the phosphorylation of BIMat Ser65 in non-neural cells can promote the degradation of BIMby the proteasome(Ley et al.,2004;Becker and Bonni,2006).Although the presence of Pin1 in a neuron-specific JNK signaling complex also allows Pin1 to bind with BIM,Pin1 could promots a conformational change in pS65-BIMand protect pS65-BIMfrom proteasomal degradation in neurons,finally leading to neuronal apoptosis (Becker and Bonni,2006;Barone et al.,2008).It should be noted that phosphorylated BIMelicits distinct effects depending on cell type,thus leading to neuronal apoptosis and the prevention of non-neuronal apoptosis(Becker and Bonni,2006,2007).In neurons,Pin1 activates the mitochondrial apoptotic machinery;this might be caused by Pin1 coming into close proximity with its substrate BIM,which is localized to the mitochondrial membrane.In non-neuronal cells,Pin1 is mainly localized in the nucleus.Pin1 and BIMare localized in distinct cell compartments;this might allow for the degradation of BIM(Becker and Bonni,2006,2007;Barone et al.,2008).
Pin1 mediates neuronal apoptosis in epilepsy
Epilepsy is a neurodegenerative disorder that is characterized by repeated seizures caused by the uncontrolled and abnormal firing of neurons (Liu et al.,2019b).Early studies indicated that the expression of Pin1 was remarkably reduced in epileptic patients (Becker and Bonni,2007;Tang et al.,2017).Studies of an epileptic mouse model suggested the interaction of Pin1 with N-methyl-D-aspartic acid receptors and that this might be associated with the excitotoxicity of pyramidal neurons (Tang et al.,2017).Excitotoxicity is a toxic event induced by an excess of excitatory amino acids receptors which leads to excitotoxic neuronal death (Liu et al.,2019b).This report was further confirmed by other researchers who found that Pin1 deletion led to a significant increase in seizure susceptibility in chemical-induced epileptic mouse models (Becker and Bonni,2007).Pin1 deletion was also shown to enhance the phosphorylation of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors by phosphorylating calcium/calmodulindependent protein kinase II (CaMKII) (Becker and Bonni,2007).In a clinical study,the levels of Pin1 were found to be reduced,while the levels of p-CaMKII and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid were significantly increased in the neocortex of epileptic patients (Tang et al.,2017).Moreover,in a mouse model of epilepsy,the re-expression of Pin1 in Pin1-deletion mice led to a significant down-regulation of p-CaMKII and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid;this was accompanied by the effective suppression of seizure susceptibility (Tang et al.,2017).Thus,Pin1 is an important controlling effector in epilepsy;this indicates that Pin1 is an attractive therapeutic strategy for epilepsy (Hou et al.,2021).
Pin1 mediates neuronal apoptosis in other neurodegenerative diseases
It has been reported that Pin plays an important role in age-related neurodegeneration.Lipofuscin is an age-associated pigment and a cytological characteristic of post-mitotic cell aging (Van Houten,2019).Hashemzadeh-Bonehi et al.(2006) found that the levels of Pin1 were progressively associated with lipofuscin in the intracellular granules of aging neurons.These researchers also found that Pin1 can be modified and become dysfunctional post-translationally.As a result,the binding of Pin1 could be cleared via the endosomal pathway as the micro-environment was not suitable for Pin1(Hashemzadeh-Bonehi et al.,2006).Eventually,this binding could reduce the amount of soluble Pin1 in neurons,thus resulting in oxidative stress;this could have a deleterious effect on neurons during aging (Hashemzadeh-Bonehi et al.,2006).
Regulated Necrosis
Under chronic neurodegenerative diseases and accident conditions,such as ischemic injury,cells may undergo morphological changes involving necrosis(Charidimou et al.,2020).Traditional views consider that necrosis is a type of passive and uncontrolled cell death that occurs around the damaged tissues.However,in recent years,some reports have indicated that,in addition to the classic form of necrosis,there is a type of procedural necrosis that can be regulated but undergoes classicical necrotic morphological changes(Degterev et al.,2005;Yuan et al.,2019).In 2005,Yuan’s team (Degterev et al.,2005;Yuan et al.,2019) first proposed a new term to define this form of RN,and referred to this as necroptosis,also known as programmed necrosis.RN is known to play vital roles in various physiological and pathophysiology processes,such as neurodegenerative disorders.Thus far,a series of different types of RN have been identified,including receptor interacting protein 3-mediated necroptosis that can be induced by tumor necrosis factor,pyroptosis that relies on the activation of caspase-1 and is triggered by microbial infection,aerastin-mediated ferroptosis that requires excessive iron,and mitochondrial permeability transition dependent RN (Yuan et al.,2019;Alu et al.,2020;Chen et al.,2020;Liao et al.,2021;Tonnus et al.,2021).Although these types of RN are named distinctly,their regulatory signaling pathways exhibit some degree of crosstalk (Alu et al.,2020).For example,in some stimuli,such as excitatory amino injury and ischemia,calcium overloading could result in different types of RN (Ros et al.,2017).However,the precise control mechanisms underlying RN are still unclear and should be further investigated.Over recent years,the relationship between Pin1 and RN has been described in certain neurodegenerative disorders;for example,Pin1 knockout mice showed significantly increased levels of retinal neuronal necrosis (Figure 3) (Kuboki et al.,2009;Wang et al.,2017,2019a).In this section,we discuss the role of Pin1 in RN and certain neurodegenerative disorders.
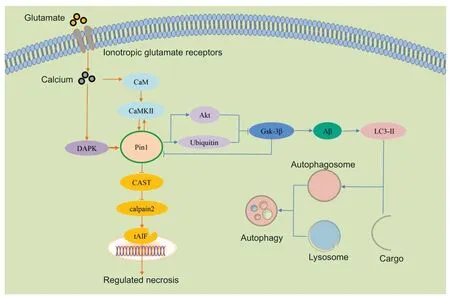
Figure 3 | The molecular mechanisms responsible for how Pin1 regulates RN and autophagy signaling pathways.
Pin1 mediates neuronal RN in brain ischemia and traumatic injury
As discussed above,Pin1 has been shown to be an important regulator of neurodegenerative diseases,such as AD.It has been suggested,that in the process of AD,DAPK1 acts up-stream of Pin1 (Bialik and Kimchi,2011).Studies have found that the over-expression of DAPK1 resulted in a strong and significant enhancement of necrotic neurodegeneration in postsynaptic neurons and that this was mediated by CaMK activation and the excessive influx of ions into the neurons (Del Rosario et al.,2015;Wang et al.,2017).Several studies have reported that DAPK1 is involved in excitotoxicity,although the regulatory mechanisms involved remain unclear.Some reports have indicated that DAPK1 acts as a modulatory adaptor to control the choice between necrosis and autophagy (Prokesch et al.,2017;Singh and Talwar,2017).However,in acute brain injury conditions,there was no significant change in the levels of light chain 3 in neurons that overexpressed DAPK1,thus demonstrating that DAPK1 may not play a role in autophagy in such acute neurodegenerative conditions (Stevens and Hupp,2008;Huang et al.,2014;Wang et al.,2017).Consistent with this result,the blockade of autophagy had no significant effects on neurodegeneration in DAPK1 knockout neurons.In this condition,DAPK1-dependent necrosis of the postsynaptic neurons could be triggered by the excessive influx of ions (Wang et al.,2017).These authors also found that Pin1 acts as an important down-regulator in DAPK1-induced excitotoxic necrosis (Wang et al.,2017).Collectively,these results suggested that DAPK1 contributes to cell necrosis but not autophagy,which is Pin1 dependent.Notably,several studies have shown that Pin1 is an upregulator of DAPK1 in neuronal development (Chen et al.,2017).This may be because the signaling cascades might be dependend on the exact scenario under different conditions (Del Rosario et al.,2015).Future studies now need to determine why the relationship between upstream and downstream of DAPK1 and Pin1 differ in a manner that is dependent on the cellular context(Ibarra et al.,2017).
Pin1 mediates neuronal RN in retinal diseases
The dysfunction of Pin1 is thought to cause retinal neuronal necrosis in glaucoma.Globally,glaucoma is the second most common eye disease that causes blindness and is characterized by the loss of retinal ganglion cells(Erickson,2021;Huang et al.,2021).Various contributors,such as ischemia,axonal transport failure,and excitatory amino acids toxicity,are thought to be major risks for the death of retinal ganglion cells.In previous studies,Wang et al.(2017,2019b) found that calpain-2 (m-calpain) played a regulatory role in the necrosis of retinal ganglion cells following glutamate excitotoxicity.Calpains are a ubiquitous form of calcium-dependent protease and have been widely investigated in different types of cellular processes,such as cell survival,proliferation,and death.Calpain-1 (u-calpain) and calpain-2,which require micromolar and millimolar concentrations of calcium for activation respectively,are the two main forms of calpains in the CNS (Wang et al.,2013,2017).Calpastatin (CAST) is an endogenous specific inhibitor of calpains (Wang et al.,2013;Cheng et al.,2018).Following glutamate excitotoxicity injury,calpain-2,but not calpain-1,is activated and cleaves apoptosis-induced factor which can then translocate to the nucleus and induce DNA degradation,ultimately resulting in neuronal RN (Wang et al.,2017,2019a).These opposing changes in calpain-1 and calpain-2 may be due to the differential roles of calpain-1 and calpain-2 after glutamate excitotoxicity (Wang et al.,2017,2019a,b).As Wang et al.(2013,2017,2019a,b) reported,calpain-1 is coupled with the synaptic N-methyl-D-aspartic acid receptor and plays a protective role after activation,while calpain-2 is coupled with extrasynaptic N-methyl-D-aspartic acid receptor and is deleterious after activation.Therefore,it is possible that the protective role of calpain-1 may be impaired after glutamate injury.In a further study,Cheng et al.(2018) showed that Pin1 could interact with and inhibit CAST in retinal neurons.Following glutamate injury,the interaction of Pin1 with CAST was enhanced,thus leading to a reductive inhibitory effect of CAST on calpain-2 (Wang et al.,2017).However,the inhibition of Pin1 expression and activation by small interference RNA and juglone (a specific inhibitor of Pin1 was shown to significantly enhance the activation of CAST;in turn,this recovers the inhibitory role on calpain-2(Wang et al.,2017).Furthermore,Wang et al.(2019a) found that Pin1 is an important down-effector of CaMKII.Following glutamate injury,Pin1 activated ionotropic glutamate receptors and led to calcium overloading (Wang et al.,2019a).Subsequently,the overload of calcium activated CaMKII and CaMKIImeditated Pin1 activation (Wang et al.,2019a).Other research reported that Pin1 can upregulate CaMKII and Pin1 expression and effectively reduce the activation of CaMKII to finally suppress seizure susceptibility (Hou et al.,2021).The upstream and downstream relationship between Pin1 and CaMKII may depend on the cellular and disease context (Hou et al.,2021).Further research should aim to specifically investigate the function of Pin1 in retinalrelated diseases.
Autophagy
Autophagy is an important physiological and pathological process that degrades intracellular dysfunctional and degenerated proteins and damaged organelles to become autophagosomes and then fuse with lysosomes to form autolysosomes (Chao et al.,2021).Under normal conditions,autophagy plays a critical role in cellular homeostasis,such as cell survival,renewal and recycling,as autophagy is the primary pathway for recycling biomolecules and organelles by degradation (Chao et al.,2021).Autophagy is also considered as a vital factor in a range of neurodegenerative conditions,such as AD (Levine and Kroemer,2008;Rami,2009;Guo et al.,2021).However,in contrast to the role of Pin1 in apoptosis,the molecular mechanisms of Pin1 regulation in autophagy signaling pathways and its role in neurodegeneration still remain unclear and need to be investigated (Figure 3
).Recent studies have revealed the underlying connections between Pin1-mediated autophagy and neuronal toxicity in AD (Del Rosario et al.,2015;Chao et al.,2021).It has been shown that increased levels of APP expression could cause AD.Reduced levels of Pin1 and increased levels GSK-3β,a 47-kDa isoform of the GSK3 family,were previously detected in the brains of AD patients and shown to be involved in the processing of APP processing(Del Rosario et al.,2015).It has also been reported that APP can be phosphorylated by GSK-3β,and that the activation of GSK-3β increases the production of APP and toxicity (Zhou et al.,2021).Further research showed that Pin1 could bind to the phosphorylated Thr330 motif of GSK-3β and inhibits its kinase activity in human neuronal and glioma cells (So and Oh,2015).Pin1 deficiency or deletion of the binding site in GSK-3β led to an increase in GSK-3β activity,the increased stability of APP,and the production of toxic APP,thus suggesting the important role of Pin1 in neuronal death (Del Rosario et al.,2015).In addition,the over-expression of Pin1 can promote the turnover of APP protein by binding and inhibiting GSK-3β kinase activity,thus providing a novel mechanism for Pin1 in protection against AD (Min et al.,2005;Ma et al.,2012).Akt has also been identified as an intermediary between Pin1 and GSK-3β (Kim et al.,2009).GSK-3β is a target of Akt and can be inhibited by Akt activation (Kim et al.,2009).Eventually,Pin1 could affect the levels of GSK-3β protein by regulating Akt (So et al.,2015).In addition,Pin1 has been considered as a regulator of the proteasome (So and Oh,2015).The inhibition of Pin1 restored GSK-3β expression by inhibiting proteasome degradation,and stimulated cell death via autophagy,as demonstrated by increased levels of light chain 3-II (So and Oh,2015).It should be noted that the inhibition of GSK-3β induces the activation of Pin1.Therefore,these data suggest that GSK-3β may also be a mediator for the regulation of Pin1 activity in autophagy (Chao et al.,2021).Collectively,this data suggests that Pin1-GSK-3β pathway-mediated neuronal toxicity acts via autophagy in AD.
Inhibitors of Pin1 and Potential Clinical Applications
Pin1 is a potential therapeutic target for neurological diseases (Zabłocka et al.,2021).By screening a series of compound libraries,a number of Pin1 inhibitors have been identified that could provide protective effects by inhibiting the activity of Pin1 PPIase.According to their specific effects on Pin1 PPIase domain activity,Pin1 inhibitors can be classified as either covalent or non-covalent types (Pinch et al.,2020).After binding to the Pin1 PPIase domain,covalent Pin1 inhibitors,such as Juglone and KPT-6566,can induce a covalent modification of the thiol group of cysteine residues in the Pin1 PPIase domain (Campaner et al.,2017;Bedouhene et al.,2021).Via these covalent modifications,covalent Pin1 inhibitors can irreversibly block the activity of the Pin1 PPIase domain (Pinch et al.,2020).Most of the Pin1 inhibitors are noncovalent types,such as 2-(3-chloro-4-fluoro-phenyl)-isothiazol-3-one (TME-001),diethyl-1,3,6,8-tetrahydro-1,3,6,8-tetraoxobenzol-phenanthroline-2,7-diacetate (PiB),all-trans retinoic acid (ATRA),and arsenic trioxide (ATO) (Mori et al.,2011;Kozono et al.,2018;Bedouhene et al.,2021).Non-covalent Pin1 inhibitors can also bind to the Pin1 PPIase domain,but inhibit Pin1 activity in a competitive manner (Kozono et al.,2018).However,the Pin1 inhibitors identified thus far are associated with limitations that restrict their use in clinical treatments (Additional Table 2
).Highly specific and potent Pin1 inhibitors are urgently needed.Juglone was the first identified inhibitor that can inhibit the activity of Pin1.Juglone has been shown to inhibit neuronal damage in some experimental models of disease,such as PD and AD.Juglone irreversibly inhibits Pin1 catalytic domain PPIase activity at high concentrations Although the inhibition of Pin1 by Juglone is effective against neuronal damage bothin vitro
andin vivo
,the lack of Pin1 specificity and toxicity at high concentrations create significant limitations with regards to potential application in clinical treatments.Therefore,further research now needs to evaluate or design potent and specific Pin1 inhibitors for therapeutic use.KPT-6566 is known to degrade Pin1 with higher levels of specificity and can reduce the expression of cyclin D1.Furthermore,KPT-6566 can induce cell apoptosis by generating ROS and inhibits cell proliferation in both breast and prostate cancer.More importantly,KPT-6566 inhibits the growth of cells over-expressing Pin1 overexpressing but not Pin1 knockdown cellsin vitro
orin vivo
,thus suggesting that KPT-6566 can specifically suppress the activity of Pin1.However,further experimental and clinical investigations are now needed to explore the efficacy and safety of KPT-6566 for the treatment of neurological diseases.TME-001,PiB,epigallocatechin-3-gallate,dipentamethylene thiuram monosulfide,6,7,4′-trihydroxyisoflavone and 4,6-bis (benzyloxy)-3-phenylbenzofuran have been found to inhibit the growth of cancer cells by inhibiting the Pin1 PPIase catalytic domain in a competitive manner (Tatara et al.,2009;Mori et al.,2011;Lim et al.,2017;Fan et al.,2019;Bedouhene et al.,2021;Della Via et al.,2021).It should be noted that positive PiB uptake in the cerebral cortex has been detected,thus indicating that PiB may be useful for the treatment of neurological diseases (Antonelli et al.,2016;Liu et al.,2019a).Epigallocatechin-3-gallate (EGCG) induces the death of cancer cells through different pathways,such as the release of nitric oxide,and is not limited to Pin1-mediated pathways (Della Via et al.,2021).In addition,only PiB has been reported to be involved in synaptic transmission by inhibiting Pin1 activity in the CNS (Antonelli et al.,2016).A previous study found that 5′-nitro-indirubinoxime can reduce the expression of Pin1 and induce G1 phase cell cycle arrest,finally inhibiting the growth of lung cells (Yoon et al.,2012).5′-Nitro-indirubinoxime can also block Polo-like kinase 1 signaling (Yoon et al.,2012).API-1 is a newly detected form of Pin1 inhibitor that can strongly inhibit the PPIase domain.Previous research found that hepatoma carcinoma cells were not affected by API-1,possibly may due to low efficacy (Sun et al.,2019).Both ATRA and ATO have been approved by the U.S.Food and Drug Administration for the treatment of patients with acute promyelocytic leukemia (Kozono et al.,2018;Bedouhene et al.,2021).Furthermore,ATO and ATRA exert a synergistic effect to inhibit the growth of breast cancer cells.ATRA and ATO can bind to the Pin1 catalytic PPIase domain in a noncovalent form,degrade the levels of Pin1,and reduce the expression of cyclin D1 (Kozono et al.,2018).However,the short half-life of ATRA may reduce its efficacy against tumors (Bedouhene et al.,2021).Moreover,the lack specificity of Pin1 also impede its application,as ATO can directly degrade cyclin D1 (Kozono et al.,2018).Although these Pin1 inhibitors are effective against proliferation in various cancer cellsin vitro
,their efficacy for the treatment of neurological diseases has yet to be investigated.Moreover,the poor solubility and specificity of these inhibitors also limit their potential therapeutic ability in the clinic.Conclusion
RCD,including apoptosis,autophagy,and RN,is a type of controlled cellular death that contributes to the physiological balance in cell clearance and organ homeostasis.Over the past few years,several studies have indicated that the aberration of Pin1 expression is a pathological characteristic and plays a crucial regulatory role in neuronal RCD,especially in apoptosis.Pin1 dysfunction also occurs in patients with various neurodegenerative diseases and is correlated with poor therapeutic efficiency.The inhibition of Pin1 plays a vital role in neuronal survival.Therefore,Pin1 may represent a potential key therapeutic target for related neurodegenerative diseases.Intriguingly,Pin1 acts in both protective and harmful roles to regulate the neuronal death;the precise effects are dependent on different cellular contexts.This is because Pin1 mediates multiple mechanisms by binding with different targets.Then,Pin1 regulates different cellular physiological processes,finally inducing different consequences.The regulatory pathways that mediate Pin1 targets are quite distinct,thus indicating the necessary of using different disease models to investigate disease development and the pharmacological treatment of Pin1-related CNS diseases.Further research is also needed to investigate the potential effect of Pin1 as a biomarker for neurodegenerative diseases and in different stages of disease.Furthermore,although many Pin1 inhibitors have been identified that can inhibit the growth of tumor cells inin vitro
,there are obvious limitations with regards to their clinical application,including poor solubility,side effects,specificity,and the lack of investigations in the CNS.Highly specific and potent Pin1 inhibitors now needed to be tested in the CNS.Furthermore,effective methods for Pin1 inhibitor screening are also needed to select potentially useful compounds.For example,Tatara et al.screened chemical compounds containing fused aromatic rings that inhibited Pin1 PPIase activity and identified PiB as an effective inhibitor for Pin1 (Tatara et al.,2009).Molecular modeling by computer simulation technology is also important as this could easily reveal the underlying interactions of the active domain of Pin1 with specific inhibitors.Furthermore,efforts should be made to promote the efficacy of combined targeted therapies and to prevent drug resistance.It should be noted that we must identify specific inhibitors with fewer adverse effects.Finally,we must also develop a highly efficient delivery system to improve the bioaccessibility and bioavailability of Pin1 inhibitors in both pre-clinical and clinical studies.It should also be noted that the signaling pathways of Pin1 in different neurodegenerative diseases is complicated and that these pathways may interact with each other.Future studies are needed to investigate such interactive networks and gain a better understanding of Pin1-mediated diseases.Remarkably,although many research studies have reported that Pin1 is an important target in apoptosis for the treatment of neurodegenerative diseases,such as AD,there are still many challenges.For example,we still need to investigate the specific role of Pin1 in RN and autophagy.
Author contributions:
Design:SCW,KX;data search,and manuscript and images preparation:SCW;manuscript and images preparation assistance and review advision:XMH.All authors revised the manuscript,and read and approved the final version of the manuscript for publication.
Conflicts of interest:
The authors declare that there is no potential conflict of interest.
Editor note:KX is an Editorial Board member of Neural Regeneration Research.He was blinded from reviewing or making decisions on the manuscript.The article was subject to the journal’s standard procedures,with peer review handled independently of this Editorial Board member and their research groups.
Availability of data and materials:
All data generated or analyzed during this study are included in this published article and its supplementary information files.
Open access statement:
This is an open access journal,and articles are distributed under the terms of the Creative Commons AttributionNonCommercial-ShareAlike 4.0 License,which allows others to remix,tweak,and build upon the work non-commercially,as long as appropriate credit is given and the new creations are licensed under the identical terms.
Additional files:
Summary of Pin1 changes in neurodegenerative diseases.
Summary of Pin1 inhibitors.

- 中国神经再生研究(英文版)的其它文章
- Neuroaxonal and cellular damage/protection by prostanoid receptor ligands,fatty acid derivatives and associated enzyme inhibitors
- Extracellular vesicles in Alzheimer’s disease:from pathology to therapeutic approaches
- Molecular approaches for spinal cord injury treatment
- Sex-biased autophagy as a potential mechanism mediating sex differences in ischemic stroke outcome
- Adipose tissue,systematic inflammation,and neurodegenerative diseases
- Interleukin-1:an important target for perinatal neuroprotection?