
邻苯二酚/微纤维胶原止血水凝胶的制备及其性能研究
2022-05-17 04:08祁增华
医疗卫生装备 2022年4期
祁增华,李 钒,丁 晟,杨 焜,田 丰
(军事科学院系统工程研究院卫勤保障技术研究所,天津 300161)
0 引言
外伤出血是战现场最常见的伤情,不可控失血是导致伤员现场死亡的首要原因。即使失血伤员后送至医院抢救,院前大量失血仍会造成较高的死亡率和严重的并发症(如截肢等)[1]。因此,研究一种能够快速、高效、救治火线伤员的止血器材意义重大。
由于环境的特殊性,海战伤员的伤口极易接触海水,而海水具有低温、高渗、高钠、高氯、偏碱性以及含菌量高等极特殊的理化特性[2]。一旦伤员受伤后伤口不及时处理经海水浸泡,就会使伤员的伤情变得更加复杂,并且长时间浸泡海水会给伤员全身都带来非常严重的伤害。目前我军应用的创伤急救敷料、急救止血绷带、自黏弹性绷带等包扎止血器材均无法满足高湿环境下的部队保障需求,因此亟须研制一种可在高湿环境下使用的具备高效止血功能的材料,进而为研制高湿环境下使用的包扎止血系列装备奠定基础。
海洋生物种类丰富多样,是开发生物材料的天然宝库,许多海底生物所具有的特殊性能也赋予了研究学者诸多的研究灵感。尽管海岸边海浪不断,但是海洋贻贝依然表现出了强大的湿黏附能力。贻贝黏附蛋白中的邻苯二酚基团是其拥有湿黏附能力的关键。受贻贝黏附蛋白的启发,研究人员研制了各种类型的聚合物模拟物,如聚乙烯亚胺-邻苯二酚、壳聚糖-邻苯二酚以及其他相关的邻苯二酚聚合物[3]。
邻苯二酚基团来源广泛,是一种多功能单体,常用于合成黏合剂和涂料。邻苯二酚及其衍生物与有机或无机基质相互作用近年来吸引了学者的广泛关注[4]。Hofman 等[5]合成了具有接枝儿茶酚基团的聚氧杂环丁烷共聚物,邻苯二酚基团占15.5%(摩尔比)的聚合物对磨砂不锈钢基体展示出5.59 MPa 的超强黏附作用,但是其潮湿表面的黏附能力显著降低。Matos-Perez 等[6]合成了聚(3,4-二羟基苯乙烯-苯乙烯)共聚物,其在干燥表面黏合强度达到11 MPa,但是水下黏合强度仅为0.3 MPa。Lee 等[7]和Kahn 等[8]认为含有邻苯二酚的分子很容易被氧化为活性醌,活性醌可以发生自聚反应形成交联产物。除此之外,邻苯二酚基团也可以与胶原相互作用,其常常被引入胶原中以改善胶原性质。
胶原是一种细胞外基质蛋白,广泛存在于皮肤、骨骼、肌腱和血管等组织中。胶原分子可以自身交联形成网络,形成的网络限制了溶剂的运动,导致胶原蛋白溶液可以转化为水凝胶。胶原水凝胶具有高度膨胀的三维网络结构,可以有效地负载生物活性分子和传递质量。并且胶原水凝胶具有免疫原性低、生物活性好等优点,常用作各种组织器官的药物或组织工程给药系统。然而,胶原水凝胶的一些缺点,如机械强度差、降解率快和热稳定性低限制了胶原水凝胶的发展和应用。Koob 等[9]使用二邻苯二酚去甲二氢愈创木酸交联胶原纤维,使胶原纤维的刚度从1 MPa 增加到约582 MPa,其力学性能与正常肌腱相当。Luo 等[10]构建了一种儿茶酚/胶原共聚物膜,用于装载一氧化氮供体。然而,关于儿茶酚及其衍生物修饰的胶原水凝胶的研究较少。Zhu 等[11]用聚多巴胺对胶原水凝胶进行了改性,但结果并不令人满意,胶原水凝胶的弹性模量仅提高到250 Pa。Duan 等[12]结合戊二醛在增强胶原性质方面的高效性,发现邻苯二酚衍生物修饰的胶原能获得具有理想性质的水凝胶。其将3,4-二羟基苯甲醛(3,4-dihydroxybenzaldehyde,DB)加入胶原蛋白溶液中,然后用高碘酸钠(NaIO4)将DB 的邻苯二酚基团氧化成醌,活性醌在胶原分子间聚合并形成交联网络,进而形成胶原水凝胶。研究结果表明,添加DB 使水凝胶的形态更加致密,抗酶性显著增强。此外,细胞增殖试验结果表明,胶原水凝胶与DB 具有良好的生物相容性。这些数据表明,这种邻苯二酚衍生物修饰的新型胶原水凝胶具有巨大的应用潜力,但是邻苯二酚衍生物修饰胶原的水凝胶在止血方面的研究还未被探索。
基于此,本研究拟利用贻贝仿生技术,模拟贻贝黏附蛋白,以3,4-二甲氧基苯乙烯(3,4-dimethoxystyrene,DMS)和4-乙烯基苯甲酸(4-vinyl benzoic acid,VBA)为原料合成邻苯二酚共聚物P(DHS-co-VBA)。用聚苯乙烯骨架代替黏附蛋白中的聚酰胺链,在骨架上接枝邻苯二酚来模拟黏附蛋白中的3,4-二羟基苯乙烯湿黏附基团。之后与微纤维胶原进行结合,探索通过仿生技术提升胶原的组织黏附性能,检测复合材料的生物相容性,并通过体外凝血检测、动物止血模型进一步评价其在高湿环境的止血效果[13]。
1 材料与方法
1.1 实验材料和仪器
实验材料包括DMS、VBA、过硫酸钾(K2S2O8)、二氯甲烷(CH2Cl2)、氯化钠(NaCl)、氯化氢(HCl)、三溴化硼(BBr3)、二甲基亚砜(dimethyl sulfo-xide,DMSO)、氯化钙(CaCl2)和甲醇(CH4O),均来自阿拉丁公司。
实验仪器包括凝胶渗透色谱仪(GPC 1515,Waters,美国)、核磁共振波谱仪(AVANCE 400,BRUKER,德国)、傅里叶红外光谱分析仪(Nicolet 380,Thermo,美国)、表面张力测量仪(OCA20,Dataphysics,德国)、微机控制电子万能试验机(C42.503,美斯特,中国)、低温恒温反应浴槽(XHDHJF-8002,霄汉,中国)。
1.2 样品制备
由于邻苯二酚基团中的羟基十分活跃,在制备过程中首先通过甲氧基将邻苯二酚基团中的羟基保护起来,随后对甲氧基进行脱保护,得到实验所需要的羟基。
(1)聚[(3,4-二甲氧基苯乙烯)-共聚-4-(乙烯基苯甲酸)][P(DMS-co-VBA)]制备。
首先利用固相萃取柱将VBA 中多余的阻聚剂去除。将1 g DMS 和5 mL VBA 加入装有60 mL 蒸馏水的三口烧瓶中进行乳液聚合反应。反应物加热至60 ℃后,将64 mg 引发剂K2S2O8溶解在2 mL 水中后滴加至三口烧瓶中,并在氩气保护的氛围下于60 ℃搅拌24 h 后加入1 mL CH4O 终止聚合。进一步加入5 g NaCl 破乳沉淀,通过离心得到固体不溶物[14],再使用CH4O 溶解后离心得到沉淀。此操作重复2~3次后,将得到的沉淀通过冷冻干燥剂除去多余的液体后,得到P(DMS-co-VBA)粉末。P(DMS-co-VBA)合成示意图如图1 所示。
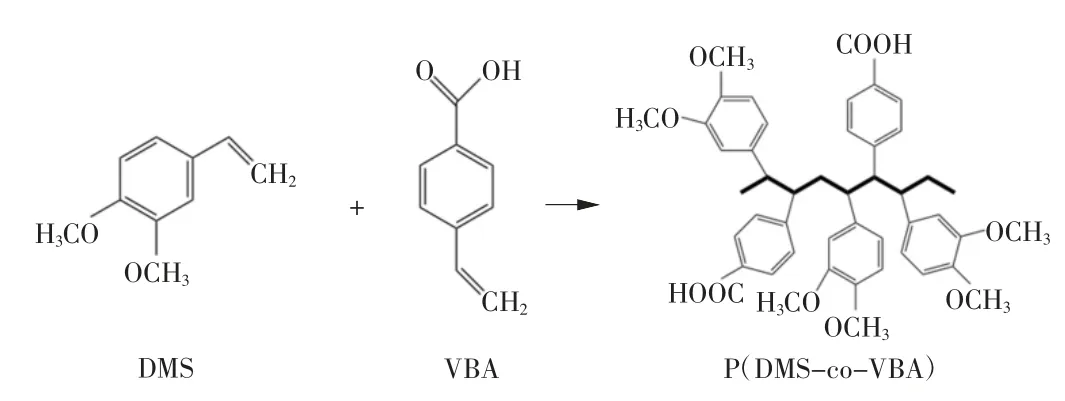
图1 P(DMS-co-VBA)合成示意图
(2)P(DHS-co-VBA)的制备。
将2 g P(DMS-co-VBA)粉末溶解在50 mL CH2Cl2中。在氩气保护的氛围下,将反应温度冷却至-10 ℃,维持10 min,然后在10 min 内逐滴加入3 mL BBr3,并不断搅拌。用300 mL 的1%HCl 分3 次重复洗涤处理该聚合物,然后在80 ℃烘箱干燥得到共聚物P(DHS-co-VBA)固体颗粒。P(DHS-co-VBA)合成示意图如图2 所示。

图2 P(DHS-co-VBA)合成示意图
(3)共聚物/胶原水凝胶制备。
将20 mg 的P(DHS-co-VBA)固体颗粒溶解在DMSO 与去离子水的混合溶剂中(体积比1∶1),加入一定质量的微纤维胶原,搅拌共混,然后在80 ℃烘箱蒸发部分溶剂,制得P(DHS-co-VBA)/微纤维胶原水凝胶。
1.3 材料物化性能的表征
1.3.1 凝胶渗透色谱(gel permeation chromatography,GPC)
首先将待测样品提前1 d 用氘代DMSO 溶剂溶解,之后将柱温设置为40 ℃、检测室的温度设置为40 ℃,用普鲁士多糖作为标准物质,将示差检测器的流速设置为1 mL/min 后,用凝胶渗透色谱仪进行检测。
1.3.2 核磁共振(nuclear magnetic resonance,NMR)氢谱
使用干燥、清洁、高质量的核磁管,以避免污染样品及匀场过程中引入不必要的麻烦。对样品进行充分的干燥和提纯,避免杂质峰掩盖有用信号,同时样品中不能混有磁性杂质,否则会扭曲磁场而降低谱峰的分辨力。用5~10 mg 固体样品盖住核磁管底部后,用氘代DMSO 试剂0.5 mL 进行溶解,氘代试剂溶解后用核磁共振波谱仪进行氢谱的测试,并扫描16 次得到NMR 氢谱。
1.3.3 傅里叶变换红外光谱(Fourier transform infrared spectroscopy,FTIR)
使用溴化钾压片法,先将样品置于80 ℃干燥箱充分干燥后,取少量样品以1∶100 的比例与溴化钾在研钵中混匀后置于不锈钢压模中压片成型,之后采用傅里叶红外光谱分析仪对样品进行红外光谱分析,以分辨力0.09 cm-1、扫描范围4000~400 cm-1观察样品对波谱的吸收情况。
1.4 亲疏水性检测
打开接触角测试仪和相连接的计算机,滴液量尽量控制在1~5 μL 以内,因为滴液过多,液滴受重力影响会产生变化,不是一个正规的圆形或者椭圆形,影响测试结果。将样品置于样品台上,需根据实际操作情况调节光源明暗度,太亮会导致液滴外轮廓不清晰,太暗则导致液滴变大或者周围太多黑点,影响测试数据的准确性。打开软件后进行自动滴液,然后上升样品台接液基线位置,原则上一键式测量是自动找出基线进行测试,特殊情况无法识别出基线的,需要操作人员手动找寻基线。接液完成后,完成水接触角的测试。
1.5 样品干湿黏附强度分析
以玻璃作为干表面的黏附基底、浸水猪皮作为湿组织表面的黏附基底,从市场购买聚氯乙烯(polyvinyl chloride,PVC)型和环氧树脂型黏合剂,通过设计搭接剪切对比实验测试P(DHS-co-VBA)/微纤维胶原水凝胶的黏附强度[5],具体操作如下:
截取2 块尺寸为70 mm×25 mm 的黏附基底,将0.5 mL(0.1 g/mL)的胶黏剂溶液涂在一块基底上。然后将涂有胶黏剂的部分搭接在一起,轻轻揉搓使混合均匀(搭接面积为25 mm×25 mm)。24 h 后对涂有胶黏剂的基底进行搭接剪切测试,拉伸速率为2 mm/min,直到2 块基底被拉开[15]。搭接剪切强度的计算公式为a=P/(b×c),式中,a 为搭接剪切强度(MPa);P 为最大拉伸作用力(N);b 和c 分别为搭接区域的长度和宽度(mm)。
1.6 样品细胞毒性测试
1.6.1 细胞增殖率
利用小鼠成纤维细胞进行毒性试验,设计磷酸盐缓冲液(phosphate buffered saline,PBS)对照组及4组P(DHS-co-VBA)/微纤维胶原水凝胶样品实验组,并选用细胞毒性实验中常用的2 个浓度进行试验。其中实验组1 为10 mg/mL 的P(DHS-co-VBA)样品(与细胞孵育后浓度),实验组2 为10 mg/mL 的P(DHS-co-VBA)/微纤维胶原水凝胶样品,实验组3为50 mg/mL 的P(DHS-co-VBA)样品,实验组4 为50 mg/mL 的P(DHS-co-VBA)/微纤维胶原水凝胶样品。每组设置4 组平行实验进行测试[16-18],具体操作如下:
(1)将1×104个小鼠成纤维细胞接种在杜尔贝科培养基(Dulbecco’s modified eagle medium,DMEM)中,并在5%CO2的培养箱内37 ℃孵育24 h。
(2)去除原培养液,将一定体积的细胞(密度为2500 个/cm2)放置于24 孔板中,共计2 板,培养基为DMEM 加入青霉素(100 U/mL)、链霉素(100 U/mL)、10%的胎牛血清。在5%CO2的培养箱内37 ℃培养24 h。
(3)加入细胞计数试剂盒(cell counting Kit-8,CCK-8),孵育4 h 后,弃去培养基,并替换为150 μL的DMSO,使用酶标仪检测细胞增殖率[19]。
1.6.2 死活细胞染色
死活细胞染色具体操作如下:
(1)将1×104个小鼠成纤维细胞接种在DMEM中,在5%CO2的培养箱内37 ℃孵育24 h。
(2)去除原培养液,将一定体积的细胞(密度为2500 个/cm2)放置于24 孔板中,共计1 板,培养基为DMEM 中加入青霉素(100 U/mL)、链霉素(100 U/mL)、10%的胎牛血清。在5%CO2的培养箱内37 ℃继续培养24 h。
(3)加入死活细胞染色剂钙黄绿素乙酰氧基甲酯(一种具有细胞膜渗透性的活细胞荧光染料,呈绿色荧光)和碘化丙啶(其仅能穿过死细胞膜的无序区域到达细胞核,嵌入细胞的DNA 双螺旋区域,并产生红色荧光)。在37 ℃水浴下孵育15 min 后,使用荧光显微镜放大50 倍捕获图像。
1.7 样品止血性能测试
1.7.1 体外全血凝固时间(CBT)检测
CBT 是一种能直观反映凝血性能的基础性检测。在新西兰兔的血液样本中加入3.8%的柠檬酸钠,实验开始时,在3 个5 mL 玻璃管中分别加入20 mg P(DHS-co-VBA)[P(DHS-co-VBA)组]、20 mg 微纤维胶原(胶原组)、10 mg 微纤维胶原与10 mg P(DHS-co-VBA)(样品组),空白对照组中不添加任何物质。然后在37 ℃水浴下孵育5 min,取1 mL 兔血与样品混合,37 ℃水浴下孵育3 min,取0.5 mL 0.025 mol/L 的CaCl2水溶液加入管中触发凝血,将试管从水浴中取出,每隔15 s 倒转1 次,直到试管中的血液不再流动[20]。
1.7.2 兔肝损伤模型止血实验
设置空白对照组、胶原实验组和P(DHS-co-VBA)/微纤维胶原水凝胶实验组,每组3 只白兔。空白对照组不添加任何止血材料,胶原实验组微纤维胶原用量为0.2 g,P(DHS-co-VBA)/微纤维胶原水凝胶组利用注射器将1 mL 的样品注入到伤口表面,其中样品中P(DHS-co-VBA)和微纤维胶原各0.1 g。实验前1 d,所有白兔必须禁食,但是不禁水。实验前,将白兔以仰卧的形式固定在实验手术台上,肌肉注射10%的陆眠宁,用药量0.75 mL/kg;待白兔麻醉后,在腹部作备皮处理并且进行酒精消毒;随后沿中线剖腹,将兔肝组织完全暴露,用无菌纱布吸去多余的腹水及血液。在兔肝上叶取下直径约1.2 cm、深度约1 mm 的组织,形成出血创面;在伤口自然出血3 s 后,用无菌纱布吸去流出的血液,然后立即将止血材料完全覆盖于创面,如图3 所示。随后每隔30 s观察一次出血情况,用已称重的无菌纱布吸去创面周围渗血,当在止血材料表面观察不到有血液渗出,且保持30 s 仍然不再出血时,认为出血已得到控制,实验结束,并记录下止血时间[21]。取下伤口处的止血材料及无菌纱布后进行称重,根据所用敷料的原始质量和止血后质量的差值,计算失血量。
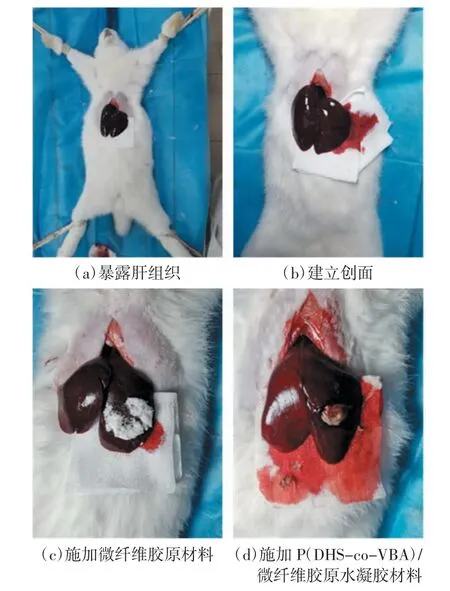
图3 兔肝损伤模型止血实验
2 结果
2.1 材料的物化性能表征结果
2.1.1 GPC
通过对图4 进行分析处理可以得到GPC 的结果,结果显示,P(DHS-co-VBA)的数均分子量为5023,重均分子量为7162,黏均分子量为6771,P(DHS-co-VBA)的多分散性指数为1.42584,分散性较好,证明聚合物已成功制备。
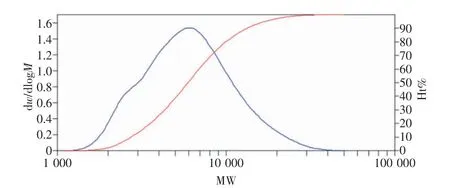
图4 P(DHS-co-VBA)分子量的分布曲线
2.1.2 NMR 氢谱
如图5 所示,NMR 氢谱与目标聚合物一致,其中化学位移ppm 6.5~8 为苯环上H 的位移,ppm 12.9左右为羧基H 位移,P(DMS-co-VBA)的NMR 氢谱结果中ppm 3.5~4 表现为甲氧基的H 的位移,而该位置的峰在图6 中P(DHS-co-VBA)的NMR 氢谱结果中消失,同时P(DHS-co-VBA)的NMR 氢谱结果同时出现酚羟基的位移峰。说明P(DHS-co-VBA)已成功脱去甲氧基,在还原的过程中,邻苯二酚基团裸露在外。
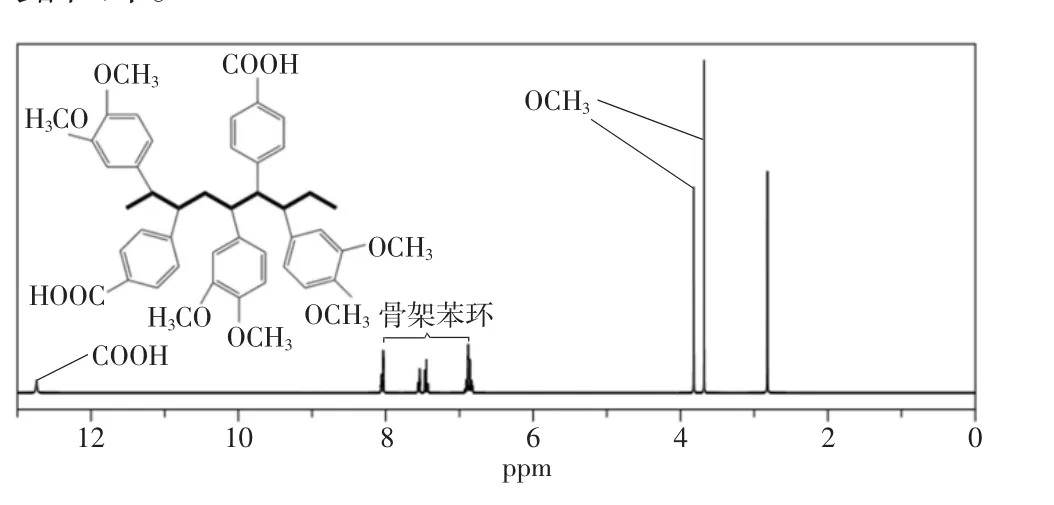
图5 P(DMS-co-VBA)的NMR 氢谱结果

图6 P(DHS-co-VBA)的NMR 氢谱结果
2.1.3 FTIR
P(DMS-co-VBA)与P(DHS-co-VBA)的FTIR图显示(如图7 所示),P(DHS-co-VBA)在1200~1300 cm-1和2500~2800 cm-1处有吸收峰,这表明P(DHS-co-VBA)共聚物表面含有大量的羧基、羟基活性基团。P(DMS-co-VBA)在2950 cm-1处的甲氧基吸收峰在还原过程中消失,也证实了邻苯二酚基团的成功制备。
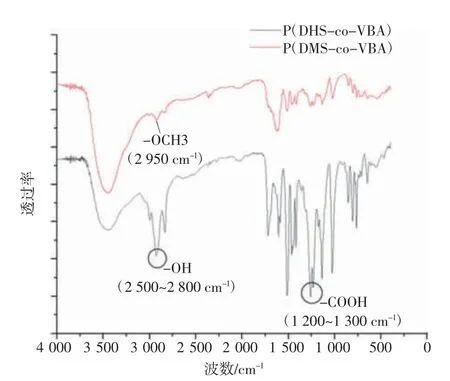
图7 P(DMS-co-VBA)与P(DHS-co-VBA)的FTIR 图
2.2 亲疏水性检测
如图8 所示,通过测试,P(DHS-co-VBA)/微纤维胶原水凝胶材料水接触角为73.5°,说明P(DHSco-VBA)/微纤维胶原水凝胶材料较为亲水,可能与P(DHS-co-VBA)表面含有大量的羧基与邻苯二酚等亲水性基团相关。

图8 水接触角检测图
2.3 P(DHS-co-VBA)/微纤维胶原水凝胶形态及性能
如图9 所示,P(DHS-co-VBA)/微纤维胶原呈水凝胶的形态,胶原因共聚物黏附力吸附在水凝胶的内部及表面,P(DHS-co-VBA)/微纤维胶原整体也有很强的黏附力,用手指拉伸一段距离后,呈丝状连接。

图9 P(DHS-co-VBA)/微纤维胶原水凝胶实物图
2.4 样品干湿黏附强度分析
由图10 可知,以玻璃为黏附基底,PVC 型黏合剂的峰值平均载荷为(44.2±3.3)N[如图10(a)所示],经过计算搭接剪切的强度为(70.7±5.2)kPa;环氧树脂型黏合剂的峰值平均载荷为(326.4±16.6)N[如图10(b)所示],经过计算搭接剪切的强度为(522.3±26.6)kPa;P(DHS-co-VBA)/微纤维胶原水凝胶样品的峰值平均载荷为(117.4±7.8)N[如图10(c)所示],经过计算搭接剪切的强度为(187.9±12.5)kPa。由图10(d)可知,P(DHS-co-VBA)/微纤维胶原水凝胶样品的黏附强度高于PVC 型黏合剂,低于环氧树脂型黏合剂。由此说明P(DHS-co-VBA)/微纤维胶原水凝胶样品在干表面具有一定的黏附强度。
由图11 可知,在浸水猪皮黏附基底黏附强度检测中,PVC 型黏合剂的峰值平均载荷为(8.6±0.8)N[如图11(a)所示],经过计算搭接剪切的强度为(13.8±1.3)kPa;环氧树脂型黏合剂的峰值平均载荷为(8.8±3.0)N[如图11(b)所示],经过计算搭接剪切的强度为(14.1±0.4)kPa;P(DHS-co-VBA)/微纤维胶原水凝胶样品的峰值平均载荷为(114.3±3.9)N[如图11(c)所示],经过计算搭接剪切的强度为(182.8±6.3)kPa。由图10(d)与图11(d)发现,制备的P(DHSco-VBA)/微纤维胶原水凝胶样品具有较强的湿黏附性能,基本与干表面的黏附强度一致,而市售的黏合剂虽然在干表面表现出很强的黏附效果,但是在湿表面表现不佳。

图10 P(DHS-co-VBA)/微纤维胶原水凝胶样品与其他样品在玻璃干表面的黏附强度测试结果
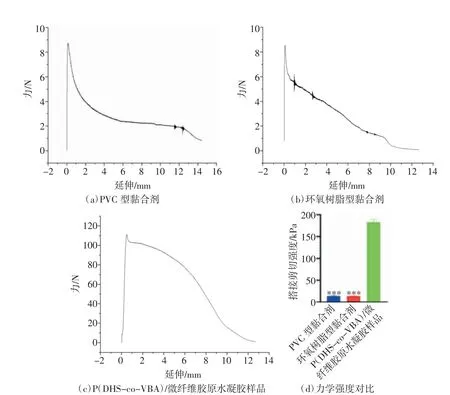
图11 P(DHS-co-VBA)/微纤维胶原水凝胶样品与其他样品在浸水猪皮表面的黏附强度测试结果
2.5 样品细胞毒性测试结果
2.5.1 细胞增殖率
如图12 所示,当细胞与检测样品孵育24 h 后,通过吸光度检测计算PBS 对照组的细胞存活率接近100%,10 与50 mg/mL 所有实验样品的细胞存活率均大于90%,说明制备的P(DHS-co-VBA)及P(DHS-co-VBA)/微纤维胶原水凝胶样品的细胞毒性低,生物相容性良好。即使在50 mg/mL 的浓度下,P(DHS-co-VBA)/微纤维胶原水凝胶材料也表现出良好的生物相容性,细胞毒性均小于2 级。

图12 CCK-8 细胞毒性检测结果
2.5.2 死活细胞染色结果
如图13 所示,死活细胞染色结果与细胞存活率测试结果一致,可以明显地看出,50 mg/mL 的P(DHS-co-VBA)及50 mg/mL 的P(DHS-co-VBA)/微纤维胶原水凝胶只有少部分的细胞死亡,同样证实了制备的水凝胶材料具有很好的细胞相容性。

图13 死活细胞染色结果(钙黄绿素乙酰氧基甲酯/碘化丙啶染色,50×)
2.6 样品止血性能测试结果
2.6.1 CBT 检测
如图14 所示,空白对照组的凝血时间为(69.0±3.6)s,P(DHS-co-VBA)组的凝血时间为(61.3±7.8)s,胶原组的凝血时间为(68.7±1.5)s,样品组的凝血时间为(63.7±5.7)s。虽然经过统计学分析发现,P(DHS-co-VBA)组、胶原组以及样品组都与空白对照组无显著性差异,但是通过凝血时间数据对比可以发现,单纯的微纤维胶原材料几乎并未改变兔血的凝血时间,而P(DHS-co-VBA)可以缩短兔血的凝血时间,同样P(DHSco-VBA)与微纤维胶原的混合物也可以缩短兔血的凝血时间,只是缩短时间的效果未达到显著差异。说明复合水凝胶具备一定的凝血效果,且较单一的微纤维胶原样品凝血效果进一步提升,说明P(DHS-co-VBA)/微纤维胶原水凝胶有助于实现血液的凝固,可能与邻苯二酚基团产生的湿黏附效果有关。
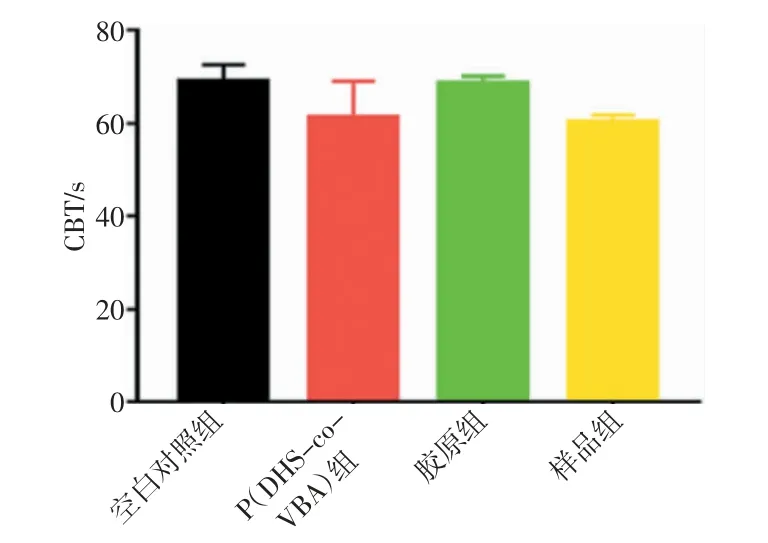
图14 CBT 检测结果
2.6.2 兔肝损伤模型止血实验
由表1、图15 和图16 可以看出,P(DHS-co-VBA)/微纤维胶原水凝胶实验组的平均止血时间为(182.7±34.8)s,平均失血量为(2.097±0.966)g;胶原实验组的平均止血时间为(191.3±27.3)s,平均失血量为(2.569±0.404)g。在止血时间方面,P(DHS-co-VBA)/微纤维胶原水凝胶实验组基本与胶原实验组一致并且远低于空白对照组;在失血量方面,P(DHS-co-VBA)/微纤维胶原水凝胶实验组远低于空白对照组,略低于胶原实验组。由于微纤维胶原在止血过程中吸收了大量的血液并凝结,在后续失血计算中难以估算材料内部吸收的血量,因此实际失血量要比可计算的失血量的数值还要大。整体而言,P(DHSco-VBA)/微纤维胶原水凝胶材料在止血性能方面优于纯微纤维胶原。
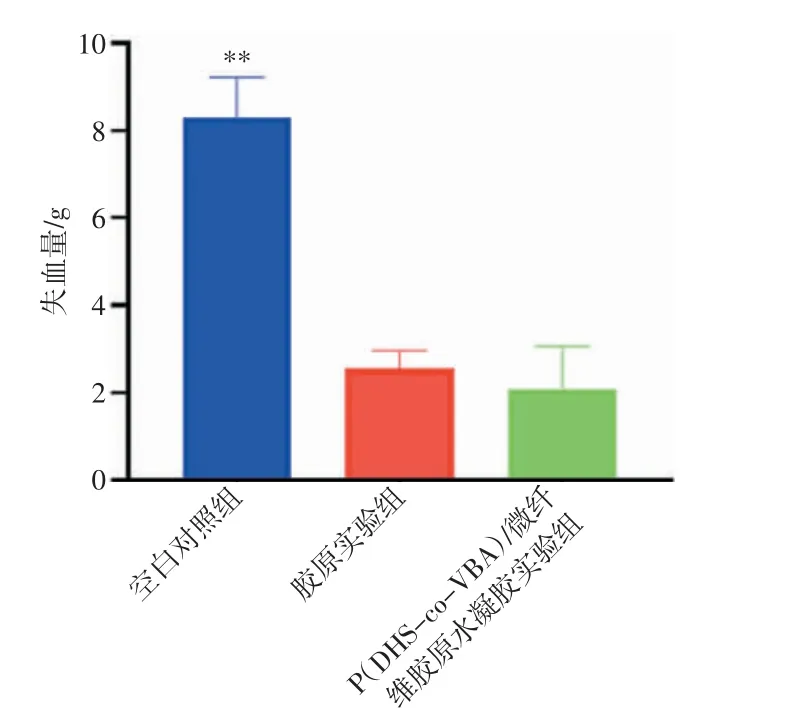
图16 各组失血量对比
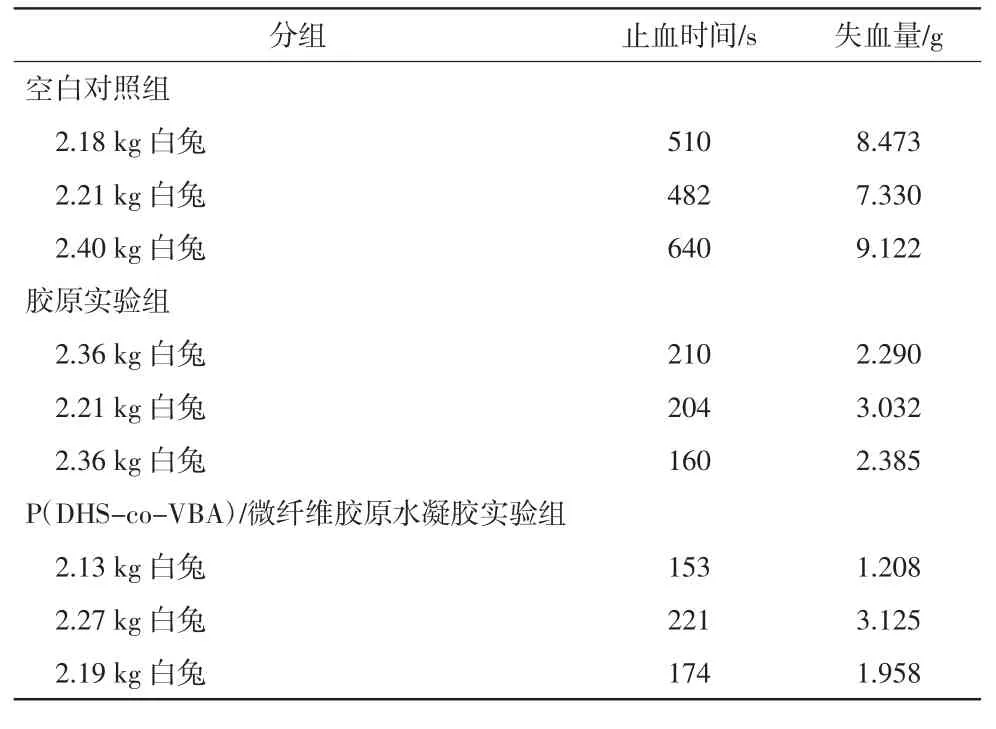
表1 各组止血时间及失血量测试结果

图15 各组止血时间对比
3 结语
本研究合成了邻苯二酚类的仿生湿黏材料,并与微纤维胶原材料复合制备了水凝胶材料,探索了邻苯二酚/微纤维胶原水凝胶材料在湿面组织的黏附强度和止血性能,并从生物相容性、体外凝血和动物创伤模型3 个方面评价了邻苯二酚/微纤维胶原水凝胶作为止血材料的可行性。研究结果表明,邻苯二酚/微纤维胶原水凝胶止血材料在高强黏附、快速止血、封闭创面方面表现出优异的性能,有望实现战创伤快速止血,进一步提升我军的卫勤保障能力。
本研究还有些许不足:(1)微纤维胶原与P(DHSco-VBA)是共混方式,并不是十分稳定,如果采用共价连接会使材料更加稳定。(2)P(DHS-co-VBA)在水中溶解性差,需将P(DHS-co-VBA)溶解在有机试剂中使用,因此会影响共聚物的细胞毒性。因此下一步的研究方向是首先制备可溶于水的邻苯二酚共聚物,并利用微纤维胶原与共聚物中的活性基团共价耦连,持续优化制备工艺,研制湿黏附强度更大的聚合材料。其次因为强大的黏附效果必然加大黏附剂在伤口处的剥离难度,因此需研制一种黏附性能可控的止血材料,实现黏附强度的精准调控。
猜你喜欢
农产品加工(2022年16期)2022-09-26
罕少疾病杂志(2022年6期)2022-06-07
皮革科学与工程(2022年1期)2022-01-15
皮革科学与工程(2021年1期)2021-03-09
小猕猴学习画刊·下半月(2020年8期)2020-07-28
恋爱婚姻家庭·青春(2019年6期)2019-06-17
少年体育训练(2018年10期)2018-11-08
军事文摘·科学少年(2018年5期)2018-09-29
现代养生·下半月(2018年7期)2018-09-12
师道·教研(2017年4期)2017-04-22
