
CO在不同氧缺陷Cu1/CeO2(110)表面的吸附:DFT + U
2022-04-22 06:17张佳松孙健伟杨建成
燃料化学学报 2022年3期
张佳松 ,王 辉,* ,王 宁 ,孙健伟 ,杨建成
(1. 哈尔滨工业大学 能源科学与工程学院,黑龙江 哈尔滨 150006;2. 河北工业大学 能源与环境工程学院,天津 300000)
与传统的NH3-SCR技术相比,CO脱硝反应(CO-SCR)具有成本低、不堵塞孔道和无二次污染等优势,因此,成为烟气脱硝领域的热点[1-3]。同时,原料的易获取也使得催化剂的研发成为CO-SCR技术的重心。其中,Cu/CeO2催化剂由于在CO还原NO方面表现出与贵金属催化剂相当甚至更优越的性能而成为了一项研究热点[4]。Zhou等[5]使用聚合物模板法制备了过渡金属铜掺杂二氧化铈微球催化剂,通过实验证明了铜掺杂氧化铈微球的催化活性明显得到了提高。戴晓霞等[6]运用原位漫反射傅里叶变换红外光谱对铜掺杂二氧化铈进行了研究,其研究结果表明,由于Cu+的存在发生了Langmuir-Hinshelwood SCR反应机理,促进了CO的有效吸附,这为铜铈催化剂极高的催化活性做出了解释。Gamarra等[7]制备了不同形貌的铜掺杂氧化铈催化剂,研究了其在过量H2中优先氧化CO的催化性能,并将其与表征结果相关联,从而揭示了CO氧化反应对二氧化铈表面分散的氧化铜实体的结构依赖性。Wong等[8]利用DFT理论对Cu单体和CuO二聚体修饰的CeO2(111)催化剂进行了研究,从电荷分离和电子转移的角度证明了Cu单体和CuO二聚体通过与CeO2之间的协同作用稳定和活化CO,从而促进CO的氧化。Chen等[9]利用DFT理论研究了铜-氧化铈界面的原子结构以及底层铜原子与还原氧化铈表面之间的电子相互作用,证实了在界面处存在部分氧化的铜物种。Cui等[10]利用DFT + U的方法研究了过渡金属Cu在CeO2(110)表面的吸附,从电荷转移的角度证明了Ce3+和Cu2+的形成。
很明显,前人的研究已经证实了铜与二氧化铈之间的相互作用可以显著提高其催化活性,并对其反应机理进行了研究。但是,需要指出的是:在真实的反应条件下,二氧化铈表面会形成不同类型的氧缺陷,而表面缺陷通常被认为是CeO2表面上各种反应的活性位[11],在催化反应中发挥着重要的作用。Nilius等[12]利用DFT理论研究了含单个氧空位的CeO2(110)表面对金原子的吸附,他们发现,金原子需要通过与氧空位附近的Ce3+进行离子交换来改变其电荷状态,这阻碍了金原子对缺陷位的占据。Jia等[13]通过DFT + U的方法研究了单个氧空位对Si掺杂CeO2(111)表面结构和电子性质的影响,结果表明,单个氧空位使表面的离子结构发生了变化,使其在费米能级附近出现了新的DOS峰。目前,很少有学者对铜掺杂氧化铈催化剂表面的不同缺陷类型进行研究报道。
吸附过程既是SCR脱硝反应中的第一步,也是重要的一步[14],有学者通过原位红外实验发现,还原剂在催化剂表面的吸附和随后的反应是形成反应中间体的重要步骤[15]。因此,吸附性能是反应能否发生的一个重要指标,对吸附性能的研究也可以评价不同缺陷表面的吸附能力。综上,本文应用DFT + U计算研究了CO在铜掺杂氧化铈(110)表面的吸附,系统研究了CO在无缺陷表面和三种缺陷类型表面的吸附性能,并对不同构型下的Mulliken电荷和电子态密度进行分析,研究结果可以为铜铈催化剂的进一步开发提供一定的理论指导。
1 计算方法与模型搭建
结构优化均采用Materials Studio 2019软件中的Dmol3模块[16],使用的交换相关泛函是广义梯度近似(GGA)中的Perdew-Burke-Ernzerhof(PBE)[17]。本研究选择的基组为双数值轨道基组加d轨道极化函数(DND),自洽场能量收敛标准为2.0 × 10-5Ha(1 Ha = 2625.4986 kJ/mol),最大力收敛标准为4 ×10-2Ha/nm,最大位移收敛标准为5 × 10-4nm。由于传统的DFT-GGA方法不能描述Ce的4f轨道电子间的强关联作用,因此,需要引入Hubbard U值进行修正以获得正确的电子结构,结合前人对CeO2的工作,本研究的计算中取U值为6 eV[18,19]。
Yang等[20]认为,对于负载金的贵金属催化剂,随着金属颗粒尺寸的减小,比活性将急剧增加。而金属粒子的大小从小体积、纳米团簇到亚纳米团簇,最终可以缩小到单个原子的大小,这就是单原子催化剂(SAC)。目前,SAC已经得到了一定的应用,有实验和理论证明SAC不仅降低了贵金属的消耗,而且具有更高的选择性、活性和稳定性[21-23],并且掺杂在氧化物表面的单个原子会表现出不同于负载型纳米粒子的独特性质[23],在此背景下,搭建了单原子铜掺杂二氧化铈催化剂,对其吸附性能进行探究。
(110)表面是低指数氧化铈表面中的第二稳定表面,化学活性更高[24]。有研究报告表明,一旦CeO2(110)表面形成氧空位,表面将更加活跃[25],因此,本研究选择对二氧化铈的(110)表面进行研究。在计算中,对于周期性(110)表面,采用了(2 × 2)的超级单元,真空层的厚度设置为1.5 nm,以避免层与层之间的相互影响。结构表层原子完全驰豫,底部四层原子固定,对CO在表层Cu原子的正上方(顶位),Cu–O键的正上方(桥位),Cu–O–Ce三原子的正上方(空穴位)等三个不同位点的吸附性能进行了研究,吸附能的计算公式如下:
式中,Eads代表吸附能;Eab代表结构优化后吸附质与吸附剂的总能量;Ea为吸附剂单独进行优化后得到的能量;Eb为吸附质单独进行优化后得到的能量。通常情况下吸附能为负值,对应于放热反应,负值越大代表吸附分子与表面间的相互作用越强[26]。如果吸附能小于-30 kJ/mol,则该相互作用归因于物理吸附;如果吸附能高于-50 kJ/mol,则归因于化学吸附[27]。
2 结果与讨论
2.1 CO在CeO2(110)表面的吸附
由于不同的计算参数会对计算结果产生不同程度的影响,因此,为了与后续的研究进行对比,首先对CO在CeO2(110)表面的吸附进行了研究。在计算时考虑了CO在CeO2(110)表面不同的吸附位点与吸附形式,所得到的吸附构型与吸附能如图1所示。从图1中可以明显看出,CO在CeO2(110)表面的最大吸附能为-38.07 kJ/mol,这说明CO在CeO2(110)表面的吸附属于较弱的物理吸附。
2.2 Cu掺杂
单原子铜掺杂氧化铈催化剂的搭建是本工作的基础,铜在二氧化铈表面的吸附位点有多个,Chen等[28]通过密度泛函理论计算发现,铜与二氧化铈表面两个氧原子进行连接时所得到的吸附能最大,结构最稳定,以此进行了铜与二氧化铈的掺杂。结构优化后得到的稳定构型如图2所示,所得到构型中Cu–O键的长度为0.178 nm,这不仅与Chen等[28]计算得到的0.181 nm数据相差不大,并且接近于立方Cu2O的实验值0.186 nm[29],这验证了本研究计算模型和方法的合理性。
2.3 CO在催化剂无缺陷表面上的吸附
根据上一节,单个铜原子通过与两个相邻氧的连接以构成掺杂,预计在CeO2(110)表面上的单个Cu原子将对CO的吸附产生重要影响。因此,首先研究了CO在无缺陷表面的吸附,考虑了顶、桥、空穴三种不同的吸附位,以及CO在表面C端吸附(C-down)与O端吸附(O-down)两种不同的吸附形式。
2.3.1 C端吸附
CO在无缺陷表面的C端吸附(C-down)如图3和表1所示,共获得三种稳定的构型,即1A、1B和1C。其吸附能分别为-161.85、-163.73和 -37.54 kJ/mol。CO在1A、1B表面的吸附属于强烈的化学吸附,在1C构型上的吸附属于物理吸附,这可能是由于CO距离活性金属Cu的距离较远所致。与2.1节中计算得到的CO在CeO2(110)表面的吸附能最大值相比,本节所得到的铜原子掺杂后的吸附能(1A、1B)明显大于该值,这证明Cu原子的掺杂显著提高了催化剂的催化活性。根据所得到的吸附构型,CO在催化剂上的最佳吸附位点是顶位。即使在1B构型中,CO的初始位置在桥位,但最终得到的吸附构型表明,CO吸附分子将自发地从桥位扩散到顶位。采用Mulliken群体分析法研究了CO与表面之间的相互作用,三种构型的转移电荷分别为0.173、0.173和0.050 |e|。吸附后的CO分子带正电荷,说明在吸附过程中电子由CO分子向底物转移,这有利于下一步化学键的形成。同时,结果也表明,构型1A和1B中分子和表面之间的相互作用更强,这与吸附能的大小一致[30]。另外,电子的转移也使得C–O键的长度发生了一定的变化。在构型1A和1B中,C–O键的长度由初始的0.114 nm伸长至0.116 nm,这也预示着下一步CO分子的解离。

表 1 无缺陷表面吸附数据(C-down)Table 1 Surface adsorption data without defects (C-down)
2.3.2 O端吸附
图4为CO在无缺陷表面的氧端吸附(O-down),其吸附能分别为-18.63(1A*)、-18.83(1B*)与-23.89 kJ/mol(1C*),与CO在无缺陷表面的C端吸附(C-down)相比,CO的氧端吸附(O-down)仅为微弱的物理吸附,图4中吸附构型的变化以及表2中的数据也印证了这一点。这说明O端吸附(Odown)并非CO在催化剂表面合适的吸附形式,C端吸附(C-down)更接近实际情况下CO在催化剂表面的吸附。Hurtado等[31]采用DFT + U的方法研究了CO在Cu掺杂WO3(110)表面的吸附,他们的研究同样表明,CO分子倾向于由C原子吸附在金属原子Cu的表面。

表 2 无缺陷表面吸附数据(O-down)Table 2 Surface adsorption data without defects (O-down)
2.4 CO在不同缺陷类型表面上的吸附
根据上一节的结论,CO更倾向于C端吸附在催化剂表面,因此,在后续的研究中仅研究了CO的C端吸附形式,构造了单原子氧缺陷、线性氧缺陷和三角形氧缺陷三种不同类型的表面,对CO在不同氧缺陷类型铜掺杂CeO2(110)表面的吸附进行研究,缺陷表面示意图如图5所示:
同样的,研究了CO在不同缺陷表面的吸附,优化后的几何构型如图6所示,不同构型的详细信息如表3所示。
CO在单个氧缺陷的Cu1/CeO2(110)表面的吸附如图6(a)所示,2A和2B是其中更为稳定的构型,其吸附能分别为-155.76和-158.16 kJ/mol。三种构型的转移电荷分别为0.172、0.174和0.033 |e|,电子从CO转移到Cu1/CeO2(110)表面。该吸附构型与上一节CO在无缺陷表面的吸附类似,结构2A和2B的吸附情况几乎相同,CO仍吸附在顶位,对于2C结构,CO在表面的吸附依旧很弱。通过与无缺陷表面的比较可以看出,单个氧原子缺陷的产生对于CO在表面的吸附影响不大。这一结果与Song等[11]的研究相似,他们采用DFT +U的方法计算了化学计量比下CO在具有单个氧空位的CeO2(110)表面的吸附,结果显示,CO在单个氧空位缺陷处的吸附依然很弱,单个氧空位的出现并没有改善空位处的表面氧或者Ce3+与CO分子的相互作用。
图6(b)展示了CO在线缺陷催化剂表面的吸附,其吸附能分别为-165.24(3A)、-230.83(3B)和-64.41 kJ/mol(3C)。氧空位产生的孤对电子在该构型中得到了显著的体现[30],3A、3B构型的转移电荷为0.057和0.099 |e|,电子仍从CO转移到Cu1/CeO2(110)表面,与之不同的是,3C构型的电子转移为-0.718|e|,这意味着CO分子吸引了吸附底物上的孤对电子[32],这可能也是CO在空穴位上的吸附行为与1C和2C构型明显不同的原因。不过,3A、3B的吸附活性依旧显著高于3C结构,从这里可以得出结论:CO在空穴位上的吸附稳定性较低。同时纵向对比表中数据可以看出,CO在线缺陷催化剂表面的吸附性能不仅在缺陷结构中表现最佳,而且高于上一节CO在无缺陷表面的吸附。Mukherjee等[33]对CeO2(111)表面的空位团簇进行了量子力学研究,结果表明,线性表面空位团簇在热力学上是更加稳定的,其不仅保证了氧的更高的迁移率,而且还提高了CeO2的储存/释放氧的能力;Esch等[34]利用DFT计算结合扫描隧道显微镜(STM)照片分析对CeO2(111)表面进行了研究,他们发现几乎所有进一步还原后形成的氧空位都是线性表面氧空位。
CO在三角形缺陷催化剂表面的吸附如图6(c)所示,三种构型的吸附能分别是-130.73(4A)、-133.54(4B)和-34.87 kJ/mol(4C),4A、4B构型的转移电荷为0.144和0.141 |e|,电子从CO转移到Cu1/CeO2(110)表面,4C构型上的转移电荷为-0.080 |e|,说明在吸附过程中有少量的电子从基底转移到CO分子[32]。另外,吸附能再度证明了CO在空穴位上的低吸附活性。在三角形缺陷催化剂表面,CO仍然倾向于吸附在顶位。同时,比较表3中的数据可以看出,CO在三角形缺陷催化剂表面的吸附活性最低,并且低于CO在无缺陷催化剂表面的吸附,这说明三角形缺陷的产生没有提高催化剂的吸附活性。其他学者对于三角形缺陷类型同样进行过研究,比如Ma等[35]对CeO2(111)表面的团簇结构进行了研究,结果表明,线性表面团簇相比于三角形表面团簇更加具备能量优势。韩仲康[36]计算了金团簇在CeO2(111)表面的吸附,结果表明,金团簇在三角形缺陷的CeO2表面的吸附能最低。
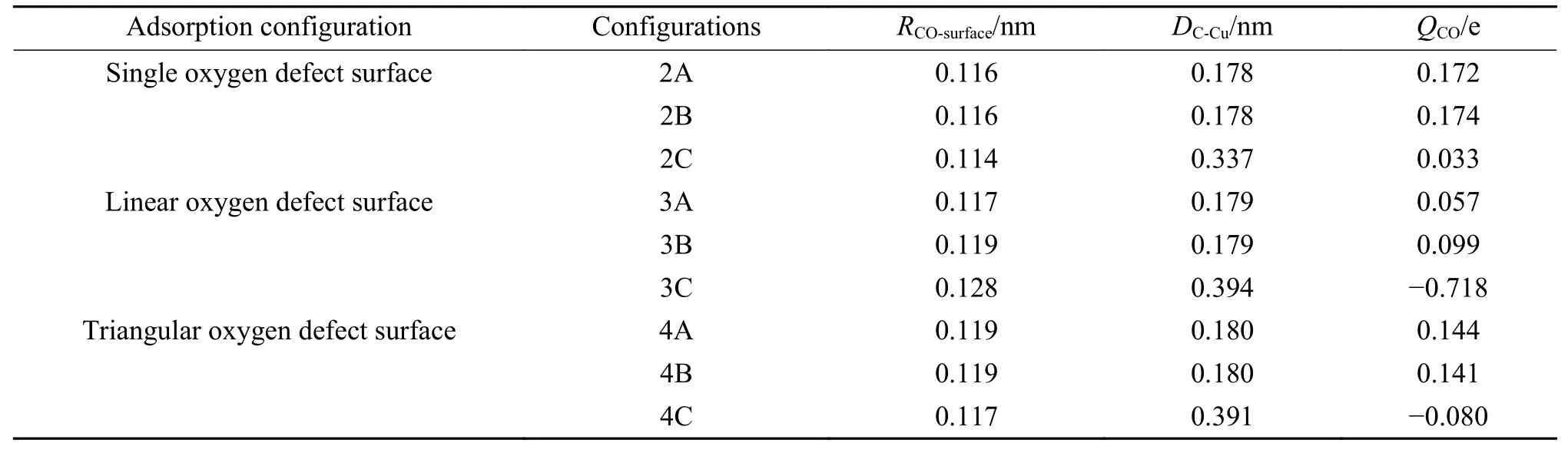
表 3 不同缺陷表面吸附数据Table 3 Surface adsorption data of different defects
2.5 不同吸附结构的电子性质
部分态密度(PDOS)的分析有助于更准确地理解气体分子和吸附基质之间的相互作用[37]。因此,为了更深入地理解和解释上一节所得到的结论,对吸附构型进行了PDOS分析。
图7展示了无缺陷吸附构型上C和Cu的PDOS,对于构型1A,C的s轨道和Cu的s轨道存在显著的相互作用,如-10.21、-7.54和-4.34 eV叠加所示。C的p轨道与Cu的d轨道也存在着相互作用,其在-7.54 eV处存在着明显的共振峰,这证实了C和Cu之间的轨道杂化[14]。1B构型与1A构型的轨道杂化情况基本一致,C的s轨道和Cu的s轨道在-10.19、-7.52和-4.38 eV处存在着轨道杂化,C的p轨道和Cu的d轨道在-7.52 eV处也存在着轨道杂化,这表明,C和Cu之间存在着强烈的相互作用。对于构型1C,从图中可以明显看出,C和Cu之间几乎不存在杂化轨道,这表明两者间的相互作用较弱,同时也解释了CO在空穴位上的弱吸附。
为了进一步探究空位缺陷带来的影响,对所研究的三种缺陷结构进行了PDOS分析。图8为单个氧缺陷吸附构型上C和Cu的PDOS,构型2A和2B中C的s轨道和Cu的s轨道存在明显的相互作用,如构型2A中-10.60、-7.93、-4.86 eV以及构型2B中-10.51、-7.99和-4.95 eV叠加所示。C的p轨道和Cu的d轨道之间也存在相同的峰,如2A中-7.93 eV和2B中-7.99 eV叠加所示,这与CO在无缺陷表面的吸附类似,表明了C和Cu之间存在着强烈的相互作用。值得注意的是,在构型2A和2B中,C的p轨道和Cu的p轨道在费米能级以上(0.81和0.82 eV)出现了新的轨道杂化峰,这意味着部分电子进入了反键轨道,使得CO分子在催化剂表面得到了活化,提高了其反应活性[38]。2C构型中C和Cu之间依旧不存在明显的杂化轨道,CO在单个氧原子缺陷表面空穴位的吸附依旧较弱。
线缺陷吸附构型上C和Cu的PDOS如图9所示,从图9中可以看出,在构型3A和3B中存在比上述几种构型更显著的轨道杂化现象。在构型3A中,C的s轨道与Cu的s轨道间存在相同的峰,如-10.66、-8.02和-5.50 eV所示;C的p轨道和Cu的d轨道也存在杂化现象,如-8.02 eV和0.57 eV处;C的p轨道和Cu的p轨道在-2.48 eV处同样存在轨道杂化现象。在构型3B中,C与Cu的s轨道杂化位于-10.38、-7.75、-5.54和-3.61 eV处;C的p轨道和Cu的d轨道在-7.75、-3.61和0.47 eV处存在轨道杂化峰。线缺陷的构造同样使C和Cu之前发生了费米能级以上的杂化,使催化剂表面的CO分子得到了进一步活化[38]。同时,与之前的两种构型不同,3C构型中CO在空穴位的吸附属于化学吸附,这归因于在-6.33 eV处C的p轨道和Cu的d轨道之间的轨道杂化。
图10展示了CO在三角形氧缺陷Cu1/CeO2(110)表面的PDOS,在构型4A中,C的s轨道和Cu的s轨道存在相同的峰,如-10.34和-7.65 eV叠加所示;C的p轨道和Cu的p轨道在-2.08和0.60 eV处存在着轨道杂化现象;C的p轨道和Cu的d轨道在-7.65和0.60 eV处也有相同的峰。在构型4B中,C的s轨道和Cu的s轨道在-10.36和-7.71 eV处存在相同的峰;C的p轨道和Cu的p轨道在-2.36和0.56 eV处也有相同的峰;C的p轨道和Cu的d轨道在-7.71 eV处存在着轨道杂化现象。同样,在三角形缺陷构型中,C和Cu之间在费米能级以上也存在着轨道杂化峰[38],这说明缺陷的构造有利于CO分子在催化剂表面的活化。在构型4C中,C和Cu之间几乎不存在杂化轨道,证明两者之间的相互作用较低。
3 结 论
本研究采用密度泛函理论(DFT)计算了CO在理想和氧缺陷Cu1/CeO2(110)表面的吸附,对吸附构型的优化结果表明,顶位是CO最稳定的吸附位,即使CO初始位于桥位,但最终仍会自发扩散到顶位,CO在空穴位上的吸附性能较弱。Cu掺杂可以显著增强CO与吸附表面的相互作用,CO解离后最有可能与Cu原子成键而被吸附。同时,在所研究的四种表面中,CO在线缺陷表面的吸附性能最佳,在单个氧原子缺陷表面和三角形缺陷表面的吸附性能要略低于CO在理想表面的吸附。进一步对PDOS的分析表明,C和Cu的s轨道杂化以及C的p轨道与Cu的d轨道之间的杂化是CO在Cu1/CeO2(110)表面吸附较强的原因。缺陷的构造使得C与Cu在费米能级以上(0.4–0.8 eV)出现了新的轨道杂化峰,这意味着部分电子进入了反键轨道,有利于CO分子在催化剂表面的活化。
猜你喜欢
无机化学学报(2022年9期)2022-09-16
军民两用技术与产品(2022年1期)2022-06-01
西北工业大学学报(2022年1期)2022-04-22
井冈山大学学报(自然科学版)(2022年2期)2022-03-31
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
校园英语·月末(2019年11期)2019-09-10
作文中学版(2018年1期)2018-11-28
北京航空航天大学学报(2017年10期)2017-04-20
读写算·教研版(2016年8期)2016-05-07
中学化学(2014年4期)2014-09-09
