
冠突曲霉flbD基因功能研究
2022-04-11 00:35余用秀葛永怡谭玉梅王亚萍刘作易
农业工程 2022年1期
余用秀,葛永怡,谭玉梅,王亚萍,邵 蕾,刘作易
(1.贵州大学生命科学学院/农业生物工程研究院,山地植物资源保护与种质创新教育部重点实验室,山地生态与农业生物工程协同创新中心,贵州 贵阳 550025;2.贵州省生物技术研究所,贵州 贵阳 550006;3.贵州省农业生物技术重点实验室,贵州 贵阳 550006;4.贵州大学农学院,贵州 贵阳 550025;5.贵州省农业科学院,贵州 贵阳 550006)
0 引言
冠突曲霉(Aspergilluscristatus)是茯茶上的优势菌,又名“金花菌”。“金花菌”的多少常被用来判断茯砖茶的品质[1]。本实验室的前期研究发现,低渗条件下,该菌以有性发育为主;在高渗条件下,冠突曲霉以无性发育为主,并产生纯的无性孢子[2]。因此,冠突曲霉是研究丝状真菌产孢机制的好材料。
丝状真菌的无性产孢是其进行繁殖的常见方式之一,在发育过程中受到了多个基因的精密调控。据报道,在模式菌构巢曲霉(Aspergillusnidulans)中,brlA、abaA和wetA组成调控构巢曲霉无性发育的中心调控途径,并决定无性产孢过程中基因激活的顺序[3]。其中,brlA位于中心调控途径的上游,该基因的激活是曲霉属产孢的一个关键步骤,主要调控分生孢子梗形成分生孢子囊的过程[4]。fluG及flbA-E是中心调控途径的上游发育激活因子,调控无性产孢的过程[5-8]。其中,flbD基因编码Myb型DNA结合蛋白,它是激活brlA基因表达所需的关键转录因子,已在酿酒酵母、构巢曲霉、烟曲霉和稻瘟病菌等真菌中有过报道,发现它们对真菌分生孢子的形成至关重要[9-13]。冠突曲霉在茯砖茶上自然生长时,在实验室低渗条件下培养时都以形成有性发育为主,这与大多数曲霉自然状态下先形成无性发育不同。因此,探索flbD基因在冠突曲霉无性产孢过程的功能,对理解丝状真菌的无性产孢机制具有重要意义。
目前,构巢曲霉(Aspergillusnidulans)及粗糙脉孢菌(Neurosporacrassa)做为模式生物开展的无性发育及无性产孢的分子调控机制研究较多,但国内外关于冠突曲霉无性产孢调控机制的报道几乎没有。因此,本研究拟以冠突曲霉(Aspergilluscristatus)为材料,期望能获得flbD基因在该菌无性发育中的功能,并初步探究冠突曲霉与模式菌构巢曲霉flbD基因功能的异同。
1 材料与方法
1.1 菌株
本试验所用的野生型冠突曲霉和根癌农杆菌AgrobacteriumtumefaciensLBA4404均来源于贵州省生物技术重点实验室,大肠杆菌EscherichiacoliDH5α感受态细胞购买于擎科生物有限公司,质粒pDHt/sk-hyg由中国科学院分子植物科学卓越创新中心王成树研究员赠送。
1.2 试剂及培养基
普通琼脂糖凝胶DNA回收纯化试剂盒、质粒提取试剂盒均购自OMEGA公司;潮霉素B、实时荧光试剂、卡钠青霉素、氨苄青霉素钠及限制性核酸内切酶等均购买于宝生物工程(大连)有限公司;引物合成由上海生工生物工程股份有限公司完成;测序由擎科生物有限公司完成;基本培养基(MM)、诱导培养基(IM)、MYA培养基参考刘逸梅等[14]进行配制;其余生化试剂均为进口或国产分析纯。
1.3 设计引物
根据Primer premier 5.0软件设计试验所需引物,如表1所示。

表1 引物信息Tab.1 Primer information
1.4 重组敲除载体的构建及转化根癌农杆菌
根据flbD基因及其上下游序列的特征,设计含有酶切位点XhoⅠ、BamHⅠ、XbaⅠ、SpeⅠ的特异性引物,用于扩增flbD基因上﹑下游侧翼序列,并将其酶切后、连接到pDHt/sk-hyg上,经验证后得到敲除载体;采用冻融法将重组敲除载体L-pDHt/sk-hyg-R转化到根癌农杆菌感受态细胞,再运用PCR扩增后测序及双酶切验证。
1.5 敲除株ΔflbD筛选及PCR鉴定
对含有重组敲除载体L-pDHt/sk-hyg-R的根癌农杆菌与野生型冠突曲霉分生孢子悬液(1×106个/mL)等体积混匀共培养,筛选转化子,用特异性引物进行PCR后验证。
1.6 敲除株ΔflbD拷贝数验证
应用5倍梯度稀释法,分别将野生型冠突曲霉基因组DNA和质粒pDHt/sk-hyg依次进行稀释,通过扩增建立GAPDH和HYG的标准曲线。对于GAPDH和HYG的检测,分别以ΔflbD、质粒pDHt/sk-hyg、野生型冠突曲霉和空白对照同时进行,每个样重复3次,根据Pfaffl法计算ΔflbD的拷贝数[16]。
1.7 ΔflbD突变株表型观察
在不同NaCl浓度的MYA培养基上,在28 ℃下培养野生型冠突曲霉和敲除株ΔflbD,应用显微镜观察菌落表型和产孢情况。
2 结果与分析
2.1 重组敲除载体构建
运用扩增上下游同源臂的引物扩增flbD基因的上、下游同源臂,长度分别为585 bp(图1a)和 857 bp(图1b)。将上述扩增片段与质粒分别进行双酶切,经验证后用T4连接酶对已酶切完全的质粒pDHt/sk-hyg和flbD上、下游同源臂进行连接,连接后的质粒用XhoⅠ和BamHⅠ、XbaⅠ和SpeⅠ分别对其进行酶切验证,电泳检测酶切的片段大小与预期一致,分别为9 317和585 bp、9 902和857 bp(图2),说明上、下游同源臂已经成功连接在质粒pDHt/sk-hyg上,重组敲除载体构建完成。
2.2 敲除载体L-pDHt/sk-hyg-R转化根癌农杆菌验证
将重组敲除质粒L-pDHt/sk-hyg-R转化到根癌农杆菌后,用特异性引物up-flbD-F and R、down-flbD-F and R进行质粒PCR验证,初步筛选得到农杆菌阳性转化子。再用XhoⅠ和BamHⅠ、XbaⅠ和SpeⅠ对农杆菌阳性转化子进行双酶切验证(图3),PCR扩增结果、酶切结果均与设计结果一致,说明重组敲除载体L-pDHt/sk-hyg-R已成功转入农杆菌中。

注:M为DL2000 DNA Marker图1 flbD基因上、下游同源臂PCR扩增结果Fig.1 PCR amplification of upstream and downstream homology arms of flbD
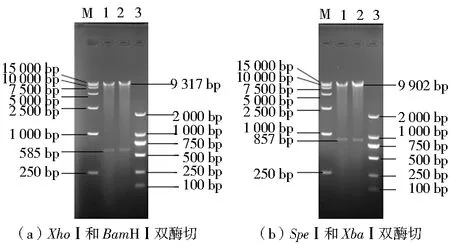
注:M为DL15000 DNA Marker;1和2为双酶切结果;3为DL2000 DNA Marker图2 flbD重组质粒L-pDHt/sk-hyg-R的双酶切结果Fig.2 L-pDHt/sk-hyg-R was digested with double enzyme

注:M为DL15000 DNA Marker;1和2为双酶切结果;3为DL2000 DNA Marker图3 来自农杆菌的重组敲除载体双酶切结果Fig.3 Results of recombination vector by double enzyme digestion
2.3 敲除株ΔflbD的PCR鉴定
根据引物设计方案(图4),利用跨潮霉素抗性基因hph的引物F1/R1进行扩增(图5a),野生型冠突曲霉扩增出2 115 bp的单一条带;阳性敲除转化子扩增出3 119 bp的单一条带。以F2/R2进行扩增(图5b),野生型冠突曲霉扩增不出特异性条带;阳性敲除转化子扩增出2 114 bp的单一条带;以F3/R3进行扩增(图5c),野生型冠突曲霉扩增不出特异性条带;阳性敲除转化子扩增出2 301 bp的单一条带。结果表明,转化子在预期位点发生同源重组,从而将目标基因敲除。

图4 引物设计方案Fig.4 Project for designing primers

图5 敲除株的PCR验证结果Fig.5 PCR verification results of the mutant
2.4 敲除株ΔflbD拷贝数验证
2.4.1 内源基因和外源基因标准曲线的建立
野生型冠突曲霉基因组DNA将作为内参标准品,含HYG基因的质粒pDHt/sk-hyg DNA作为外源标准品,通过实时荧光定量PCR扩增GAPDH和HYG的目标片段(图6a和图6b),溶解曲线均为单锋(图6c和图6d),建立了5倍浓度梯度标准曲线(图6e和图6f),GAPDH和HYG标准曲线的R2分别为0.999、0.997,扩增效率E值分别为0.933、0.963,扩增结果特异性好,可用于后续试验。

图6 内参基因和潮霉素基因片段的扩增、熔解曲线与标准曲线Fig.6 Amplification,melting and standard curves of GAPDH and HYG gene
2.4.2 目的基因拷贝数检测
对于HYG和GAPDH的扩增,分别以△flbD﹑质粒pDHt/sk-hyg﹑野生型冠突曲霉和空白对照同时进行3次重复反应。从扩增曲线和溶解曲线可以看出,△flbD中HYG和GAPDH为特异性扩增,溶解曲线为单峰(图7a和图7b);质粒pDHt/sk-hyg DNA作为GAPDH扩增的阴性对照,它的结果显示无GAPDH的扩增;野生型冠突曲霉基因组DNA作为HYG扩增的阴性对照,它的结果显示无HYG的扩增;同时,NTC(空白对照)无荧光信号。


图7 △flbD中的GADPH及HYG的扩增曲线和溶解曲线Fig.7 Amplification and melting curves of the GADPH and HYG from knockout strains
根据Pfaffl法,当N=1时,可以推断敲除菌株的拷贝数为1[16]。通过计算得到敲除株ΔflbD的N值接近于1,即敲除株拷贝数为单拷贝。
2.5 敲除菌株表型观察
敲除菌株ΔflbD和野生型冠突曲霉的菌落观察表型(图8)。结果显示:ΔflbD敲除株与野生型菌株的菌落直径和色素均没有明显差异,但敲除株的菌落呈现蓬松状,产生了大量“棉花”状气生菌丝,且菌落边缘较稀疏。在3 M NaCl的MYA培养基上培养7 d,可以看到敲除株产生的分生孢子比野生型的少,并且敲除株的分生孢子主要集中形成于菌落中心。因此,采用马跃等[17]的研究方法对含有1 mol/L和3 mol/L NaCl的MYA培养基上培养的野生型冠突曲霉与ΔflbD菌株进行分生孢子计数,结果表明野生型的分生孢子数量是敲除株的4.7倍,但子囊孢子的数量几乎没有变化(图9);其次,对野生型与敲除株的产孢结构的观察结果显示敲除株的分生孢子产孢结构的形成较野生型延迟(图10)。这表明flbD基因对冠突曲霉无性发育及分生孢子的产生了正调控作用。

图8 28 ℃培养野生型冠突曲霉(Aspergillus cristatus)与敲除株ΔflbD的菌落表型Fig.8 Phenotype of wild-type and ΔflbD strain of Aspergillus cristatus
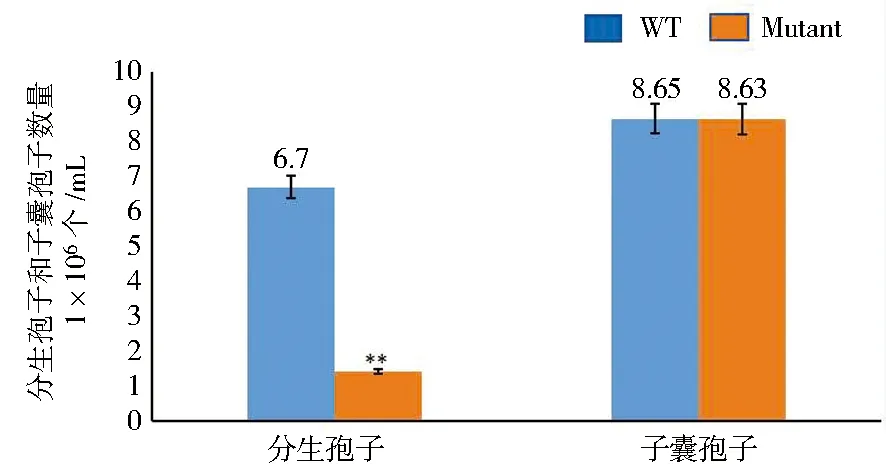
图9 敲除株ΔflbD和野生型冠突曲霉(Aspergillus cristatus)分生孢子和子囊孢子数量统计Fig.9 Statistics of conidia and ascospore from wild-type and ΔflbD strain of Aspergillus cristatus
3 讨论
本研究采用根癌农杆菌介导的同源重组法对野生型冠突曲霉中flbD基因进行敲除,然后观察flbD基因敲除株的形态变化,据此推测flbD基因在冠突曲霉发育过程中的功能。在构巢曲霉中,flbD基因是无性发育过程的重要调控因子之一,构巢曲霉中的flbD基因缺失后会导致分生孢子产生延迟及数量减少,并可能形成异常子实体[8-10]。

注:图B,F(bar:20 μm);C,E(bar:50 μm)。图10 ΔflbD突变株与冠突曲霉(Aspergillus cristatus)野生型的观察结果Fig.10 Observation results of wild type and ΔflbD strain from Aspergillus cristatus
本研究在获得flbD敲除株的基础上,通过观察发现敲除株的分生孢子少于野生型,同时在含3M氯化钠的培养基上,突变株分生孢子的产生有延迟现象,这与构巢曲霉的报道相一致[8-10]。同时,本研究结果与禾谷镰刀菌(Fusariumgraminearum)的flbD基因敲除株相比有明显差异,禾谷镰刀菌中flbD基因的缺失将阻止菌丝分化形成分生孢子梗和子囊壳,但冠突曲霉中flbD基因的缺失并不会阻断其有性及无性产孢结构的形成[18]。
构巢曲霉的fluG、flbA、flbB、flbC、flbD和flbE基因是激活brlA基因所必须的,如构巢曲霉的研究显示,flbE和flbB相互作用共同激活flbD,flbB又与激活的flbD以互作方式共同激活了brlA的转录[8]。据报道flb家族中的任何基因发生突变都会导致形成大量未分化的气生菌丝,使菌落呈现“蓬松”或棉花状外观,最终使brlA表达量大大降低,课题组前期对该菌flbA敲除后也出现了类似的表型[7,19]。本文对冠突曲霉的flbD基因进行了敲除,其敲除株的菌落也呈现蓬松状,同样产生了大量的“棉花”状气生菌丝,并且其分生孢子数量显著减少,这些现象与已报道的构巢曲霉相类似。因此,猜测冠突曲霉的flbD基因对该菌无性发育的调控方式可能与构巢曲霉类似。
4 结论
本研究通过同源重组获得了flbD基因的敲除菌株,形态学观察的结果表明冠突曲霉的flbD对该菌的无性产孢具有正调控作用,该基因的缺失将导致冠突曲霉分生孢子减少和产孢结构的形成延迟。本研究将有助于控制冠突曲霉的合适产孢方式和产孢量,从而可促进茯砖茶的产业化发展;同时将为丝状真菌的无性发育研究提供借鉴。
猜你喜欢
现代食品(2022年17期)2022-10-20
中学生物学(2022年8期)2022-10-13
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
无线互联科技(2022年2期)2022-04-20
三农资讯半月报(2020年11期)2020-06-21
江苏农业学报(2019年1期)2019-09-10
江苏农业科学(2019年11期)2019-07-22
热带作物学报(2019年4期)2019-06-11
安徽农学通报(2017年23期)2017-12-27
中国质量万里行(2015年1期)2015-01-27
