
基于二维相关红外光谱的陈皮快速鉴别研究
2022-03-30 02:09杨济齐沈婉莹魏晓芳杜文超张景照唐旭东甘肃中医药大学兰州730000深圳清华大学研究院创新中药及天然药物研究重点实验室广东深圳58000广东省创新中药及天然药物研究工程中心广东深圳58000
中南药学 2022年3期
杨济齐,沈婉莹,魏晓芳,杜文超,张景照,唐旭东,*(.甘肃中医药大学,兰州 730000;2.深圳清华大学研究院创新中药及天然药物研究重点实验室,广东 深圳 58000;3.广东省创新中药及天然药物研究工程中心,广东 深圳 58000)
陈皮为芸香科植物橘Citrus reticulata
Blanco及其栽培变种的干燥成熟果皮,具有理气健脾,燥湿化痰等功效。现代药理与化学研究表明其含有黄酮类、挥发油等活性成分,具有抗肿瘤等作用,在中成药和食品中应用广泛。陈皮以广陈皮为道地,以新会为道地核心产区。受地域等条件限制,市面上流通的广陈皮占比不大,掺假售假、以次充好问题频频发生,在一定程度上制约了陈皮产业发展。基于红外光谱三级鉴别法能够实现对陈皮的快速、无损鉴别,本实验对14 批不同产区、不同储存年限的陈皮进行二维相关红外光谱分析,为其鉴别和质量控制提供实验依据。1 材料
1.1 仪器
中药粉碎机(拜杰BJ-800A),超声仪(昆山美美超声仪器有限公司),电热套(海宁市新华医疗器械厂),水分测定管(北玻),高效液相色谱仪(Thermo),电子天平(瑞士Mettler-Toledo,精度:十万分之一),电子天平(Explorer,精度:十万分之一),Perkin Elmer FI-IR Spectrometer Frontier 傅里叶变换红外光谱仪(配备DTGS 探测器,SYD TC-01 程序控制变温附件,FW-4 型压片机,天津天光光学仪器有限公司)。
1.2 试药
14 批陈皮样品通过合作基地、药材公司购买,产区包括广东、广西、浙江、四川、湖南、湖北等地,年份为2010—2020年,其中S1 ~S8样品为广陈皮,经南方医科大学刘传明老师鉴定为茶枝柑Citrus reticulata
‘Chachi’(广陈皮);S9 ~S14 为网购陈皮药材。样品具体信息见表1。表1 陈皮样品信息
Tab 1 Citri reticulatae pericarpium sample information
编号产地年份来源S1广东省江门市新会区双水镇2020基地S2广东省江门市新会区双水镇2019基地S3广东省江门市新会区双水镇2018基地S4广东省江门市新会区天马村2015基地S5广东省江门市新会区天马村2010基地S6广东省江门市新会区梅江村2015药材公司S7广东省江门市开平市2020药材公司S8广西省2017药材公司S9浙江省2019药材公司S10浙江省2018药材公司S11四川省2020药材公司S12湖南省2018药材公司S13湖北省2020药材公司S14湖北省2018药材公司
甲苯(广州牌化学试剂),甲醇(分析纯,西陇科学),甲醇(色谱纯,avantor),溴化钾(光谱级,Alfa Aesar)。
陈皮对照药材(批号:120969-201510);橙皮苷(纯度95.3%,批号:110721-202019)、川陈皮素(纯度99.7%,批号:112055-202102)、橘皮素(纯度99.7%,批号:112054-202102)(对照品,中国食品药品检定研究院)。
2 方法与结果
2.1 陈皮水分测定
精密称定适量过二号筛陈皮粉末,参考2020年版《中国药典》,按照通则0832 第四法甲苯法对各批次陈皮进行水分测定,计算含水量(%)。结果14 批陈皮水分含量在6.49%~12.49%,均不超过13%,符合药典规定。8 批广陈皮药材水分含量在6.49%~12.39%,另6 批陈皮药材水分含量在8.22%~12.49%。
2.2 陈皮中橙皮苷、川陈皮素、橘皮素含量的测定
2.2.1 色谱条件 参照2020年版《中国药典》广陈皮高效液相色谱法(通则0512)测定橙皮苷、川陈皮素、橘皮素含量。色谱条件:Agilent Eclipse XDB-C色谱柱(5 μm,4.6 mm×250 mm),流动相为乙腈(A)-水(B)梯度洗脱(0 ~10 min,22%A,10 ~20 min,22%~48%A;20 ~35 min,48%A),柱温25℃,流速1 mL·min,检测波长:283 nm(0 ~20 min)、330 nm(20 ~35 min),进样量5 μL。
2.2.2 混合对照品溶液 精密称定对照品橙皮苷、川陈皮素、橘皮素适量置量瓶中,甲醇定容至10 mL,制成每1 mL 含橙皮苷0.2034 mg,川陈皮素25.0845 μg,橘皮素16.3348 μg 的混合对照品溶液。
2.2.3 供试品溶液 14 批陈皮经粉碎后过二号筛,精密称定0.2 g,精密加入25 mL 甲醇,超声(功率300 W,频率40 kHz)45 min,补重,摇匀,过滤,取续滤液,即得。
2.2.4 样品含量 样品测定结果见表2。8 批两广区域陈皮的橙皮苷含量在2.11%~6.93%,川陈皮素含量在0.21%~0.48%,橘皮素含量在0.23%~0.45%,3 种成分含量最高的均为2020年开平产区陈皮。另6 批陈皮的橙皮苷含量在5.27%~9.11%,川陈皮素含量在0.02%~0.70%,橘皮素含量在0.01%~0.58%。与非两广产区陈皮相比,两广产区陈皮的橙皮苷含量普遍较低,2019年浙江、2020年湖北产区陈皮所含川陈皮素、橘皮素含量很低。
表2 14 批陈皮含量测定结果
Tab 2 Content determination of 14 batches of citri reticulatae pericarpium
样品编号橙皮苷/% 川陈皮素/% 橘皮素/%川陈皮素+橘皮素/%S14.310.280.260.54 S23.380.210.230.44 S33.010.230.260.49 S43.330.280.280.56 S52.830.380.320.70 S62.110.320.270.59 S76.930.480.450.93 S82.880.250.240.49 S97.520.020.010.03 S105.270.700.581.28 S119.110.350.210.56 S126.810.650.581.23 S137.100.070.060.13 S146.250.640.551.19
2.3 陈皮红外光谱三级鉴别
2.3.1 样品制备 采用压片法制备样品:1 mg 样品加入100 mg 光谱纯溴化钾(KBr)粉末,红外灯照射下研磨均匀,压片,加压至18 ~20 MPa,维持压力3 min。样品过9 号筛、经五氧化二磷真空干燥24 h,KBr 使用前经120 ℃烘干,过200 目筛。
扫描范围4000 ~400 cm,分辨率4 cm,扫描次数64 次,测谱方式为透射谱,扣除KBr背景,实时扣除水和CO的干扰。按照每分钟2℃的升温速度对其进行加热,30 ~120 ℃温度内每隔10 ℃测试一次,得到一系列红外光谱。
2.3.2 方法学考察 取样品S1,采集室温下的红外光谱图。压片后置干燥器中,每0、1、2、3、4、5 h 测定一次,所得红外光谱图基本一致,谱间相关系数分别为1.000、0.9986、0.9976、0.9976、0.9980、0.9964,RSD
为0.12%,结果表明仪器稳定性良好。取样品S1,连续扫描5 次,所得红外光谱图基本一致,谱间相关系数分别为1.000、0.9999、0.9998、0.9998、0.9997,RSD
为0.010%,结果表明仪器精密度良好。取样品S1,采集室温下红外光谱图,重复压片5 次,所得红外光谱图基本一致,谱间相关系数分别为1.000、0.9958、0.9934、0.9815、0.9846,RSD
为0.79%,表明方法重复性良好。取陈皮对照药材,制备样品,经数据处理后得到陈皮对照药材在1700 ~1396 cm波段、1150 ~996 cm波段的二维相关红外光谱。重复实验3 次,所得谱图中相关峰位置及相对强度基本一致。结果表明该方法具有良好的重复性。
2.3.3 数据处理 谱图经基线校正、归一化后,采用Spectrum 软件的求导功能,13 点平滑,得红外二阶导数谱图。二维相关红外光谱图采用清华大学自行设计的二维相关分析软件进行处理。谱间相关系数由红外光谱仪Compare 功能计算得到。
2.3.4 红外光谱解析 图1 和图2 为14 批陈皮样品及相关对照药材、对照品的红外光谱图。以广东新会陈皮S1 样品的红外谱图为参考,计算8批广陈皮样品谱间相关系数;以陈皮对照药材为参考,计算6 批陈皮样品谱间相关系数,见表3。
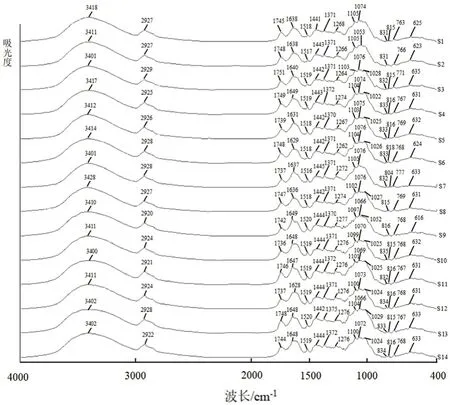
图1 14 批陈皮样品红外光谱图(4000 ~400 cm-1)Fig 1 Fourier transform infrared spectroscopy(FTIR)spectra of 14 batches of citri reticulatae pericarpium samples(4000 ~400 cm-1)
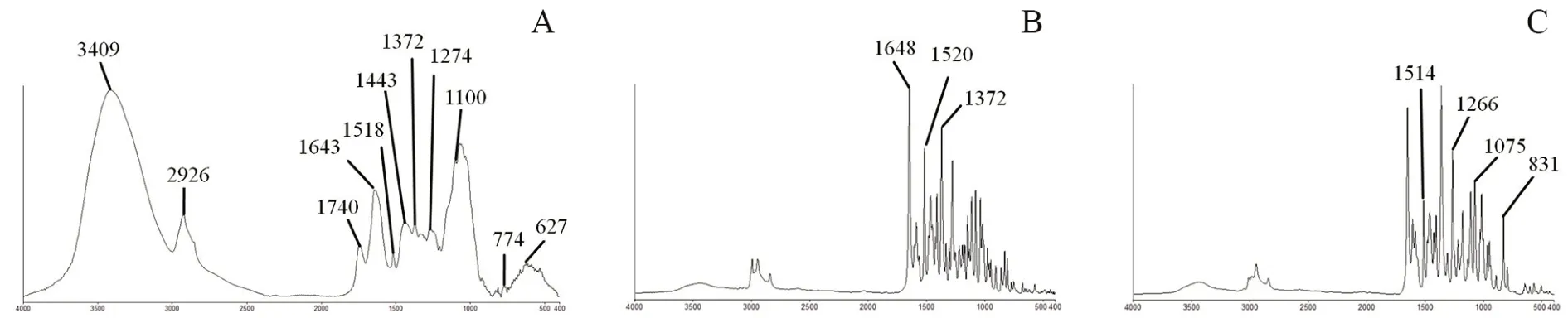
图2 对照药材及对照品红外光谱图(4000 ~400 cm-1)Fig 2 FTIR spectra of reference materials(4000 ~400 cm-1)
表3 谱间相关系数
Tab 3 Correlation coefficient between spectra
样品谱间相关系数样品谱间相关系数S11.0000 S90.8422 S20.9749 S100.9132 S30.9701 S110.9331 S40.9513 S120.9286 S50.9345 S130.8991 S60.9446 S140.8947 S70.9465陈皮对照药材1.0000 S80.9333
8 批广陈皮的红外光谱图在(3414±14)cm,(2927±2)cm,(1744±7)cm,(1640±9)cm,(1517±2)cm,(1443±2)cm,(1371±1)cm,(1268±6)cm,(1103±2)cm,(770±7)cm,(629±6)cm附近含有明显特征峰。其中(3414±14)cm附近为O-H伸缩振动峰,(2927±2)cm附近为C-H 伸缩振动峰,(1443±2)cm,(1371±1)cm附近为C-H 弯曲振动峰,(1744±7)cm,(1640±9)cm附近为C =O 伸缩振动峰。
6 批陈皮样品的红外光谱图与广陈皮相差不大,在(3405±6)cm,(2924±4)cm,(1742±6)cm,(1638±11)cm,(1519±1)cm,(1443±1)cm,(1372±3)cm,(1276±1)cm,(1100±4)cm,(767±1)cm,(624±9)cm附近含有明显特征峰。14 批样品与陈皮对照药材的红外谱图在3409 cm,2926 cm,1740 cm,1643 cm,1518 cm,1443 cm,1372 cm,1274 cm,1100 cm,774 cm,627 cm处对应;与川陈皮素对照品的红外谱图在1648 cm,1520 cm,1372 cm处对应;与橘皮素对照品的红外谱图在1514 cm,1266 cm,1075 cm,831 cm处对应。各样品在1074 cm或1053 cm附近,831 cm或815 cm附近存在吸收峰。
2.3.5 二阶导数光谱解析 1750 ~ 1400 cm波段、1150 ~985 cm波段内的二阶导数谱图(横坐标为波长,单位cm,纵坐标为吸光度)见图3 ~6。在1700 ~1450 cm,红外谱图上可以观察到(1638±11)cm,(1518±2)cm两个吸收峰,二阶导数谱中可以观察到的共有峰为(1649±3)cm,(1604±3)cm,(1519±2)cm,(1468±2)cm等4 个明显的吸收峰。与陈皮对照药材的二阶导数光谱在1649 cm,1604 cm,1519 cm,1468 cm处对应;与川陈皮素对照品的二阶导数光谱在1648 cm,1605 cm,1520 cm,1467 cm处对应;与橘皮素对照品的二阶导数光谱在1652 cm,1609 cm,1514 cm处对应。二阶导数光谱提高了谱图分辨率,与红外光谱相比可以获得更多的样本信息。红外光谱中,(1518±2)cm和(1443±2)cm间没有吸收峰,而在二阶导数光谱中,可以明显地观察到(1468±2)cm这个在红外光谱中观察不到的吸收峰。广陈皮各处吸收峰强度弱于非两广地区陈皮,表明其在含量上可能存在差别。

图3 14 批陈皮二阶导数光谱(1750 ~1400 cm-1)Fig 3 Second derivative infrared spectra of 14 batches of citri reticulatae pericarpium(1750 ~1400 cm-1)
在1150 ~985 cm波段,二阶导数光谱不仅可以观察到(1078±2)cm,(1050±2)cm等吸收峰,还可以观察到(1010±5)cm这个明显的吸收峰。与川陈皮素对照品的二阶导数光谱在1115 cm,1080 cm处对应;与橘皮素对照品的二阶导数光谱在1111 cm,1075 cm处对应。随着年限增加,产自双水镇的陈皮在1107 cm处由单峰变为双峰。

图4 对照药材及对照品二阶导数光谱(1750 ~1400 cm-1)Fig 4 Second derivative infrared spectra of the reference(1750 ~1400 cm-1)

图5 14 批陈皮二阶导数光谱(1150 ~985 cm-1)Fig 5 Second derivative infrared spectra of 14 batches of citri reticulatae pericarpium(1150 ~985 cm-1)

图6 对照药材及对照品二阶导数光谱(1150 ~985 cm-1)Fig 6 Second derivative infrared spectra of reference (1150 ~985 cm-1)
2.3.6 二维相关红外光谱解析 采用二维相关光谱对不同产区、不同储存年限陈皮药材进一步区分。图7 为陈皮对照药材及14 批样品在1700 ~1396 cm波段内的二维相关红外光谱。它们的相关峰在强度、位置、个数上存在差异。对照药材含有5 个吸收峰,最强峰位置在1604 cm处。2020年陈皮:双水产陈皮最强峰位置在1643 cm处,开平产陈皮最强峰位置在1603 cm处,四川、湖北产陈皮最强峰位置在1653 cm处。2019年陈皮:双水产陈皮最强峰位置在1404 cm处,浙江产陈皮最强峰位置在1605 cm处。2018年陈皮:双水产陈皮最强峰位置在1648 cm处,浙江、湖南产陈皮最强峰位置在1402 cm、1401 cm处。2017年广西产陈皮在1403 cm,1511 cm,1605 cm,1649 cm处有4 个明显的自动峰,最强峰在1605 cm处。2015年天马与梅江产陈皮对比结果表明:两者最强峰位置大致相同,梅江产陈皮在1400 cm处相对峰强度略强于天马产陈皮。根据这些差异,可以实现对不同产区陈皮的快速鉴别。随着储存年限的增加,天马产陈皮1595 cm处吸收峰增强,1653 cm处吸收峰减弱。湖北产区1401 cm处吸收峰变强,最强峰由1653 cm移至1605 cm处。浙江产最强峰位置由1605 cm移至1402 cm处。
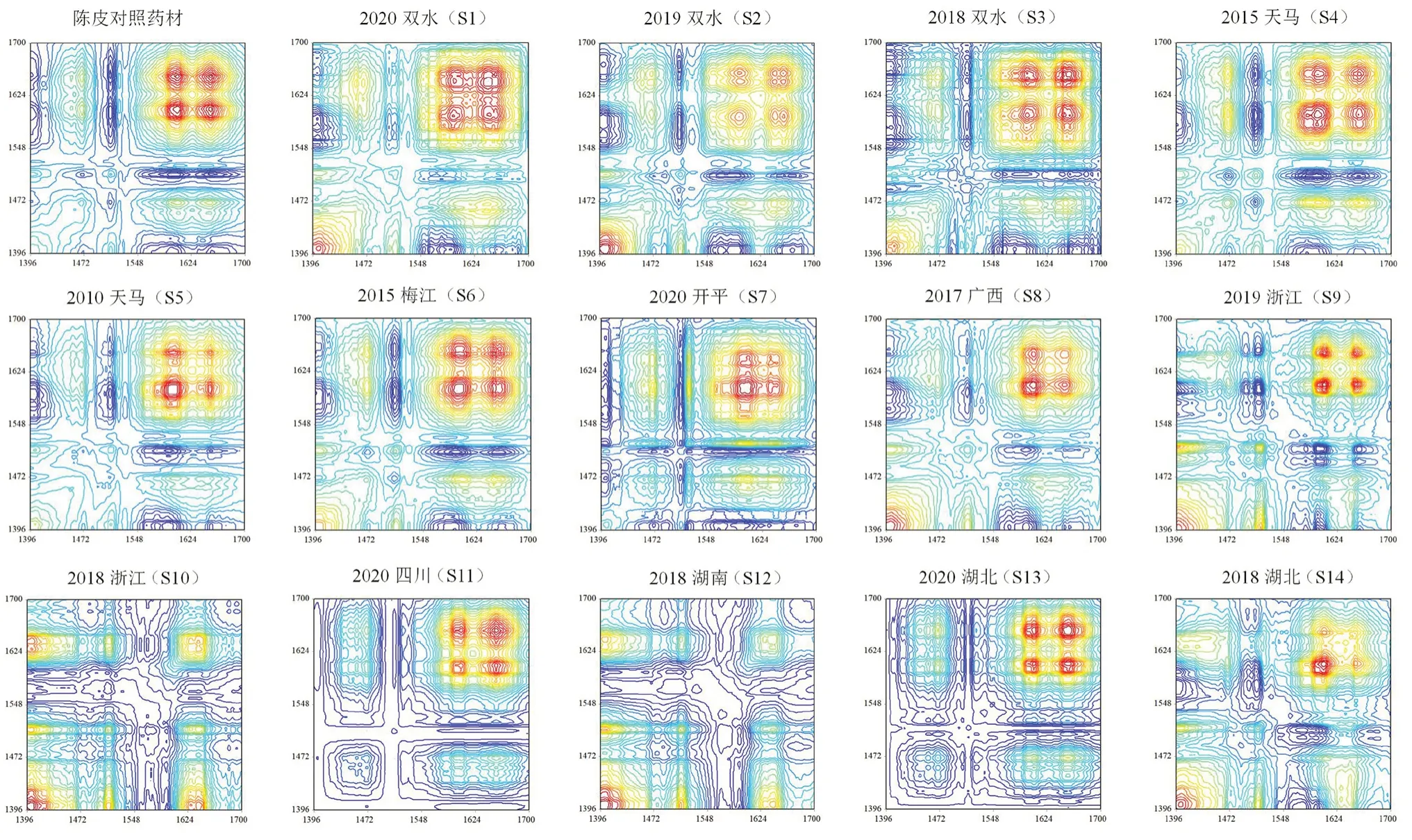
图7 陈皮对照药材及14 批样品二维相关红外光谱(1396 ~1700 cm-1)Fig 7 2D-IR synchronous correlation spectra of the reference and 14 batches of citri reticulatae pericarpium(1396 ~1700 cm-1)
进一步分析陈皮对照药材及14 批样品1150 ~996 cm波段二维相关红外光谱,如图8所示。在该范围内,不同产区陈皮自动峰、最强峰的位置及峰的相对强度有所差异。对照药材含有5 个吸收峰,最强峰位置在1001 cm处。广陈皮的二维相关谱图形成非常明显的4×4 矩阵,含有5 个自动峰。2020年陈皮:双水、四川产陈皮最强峰位置在1094 cm、1092 cm处,开平产陈皮最强峰位置在1004 cm处,湖北产陈皮最强峰位置在998 cm处。2019年陈皮:双水产陈皮最强峰位置在1042 cm处,浙江产陈皮最强峰位置在1090 cm处。2018年陈皮:双水产陈皮最强峰位置在1042 cm处,浙江、湖南产陈皮最强峰位置在1090 cm、1092 cm处。2017年广西产陈皮在1002 cm,1044 cm,1066 cm,1092 cm,1133 cm处有5 个明显的自动峰,最强峰在1044 cm处。2015年天马与梅江产区陈皮对比结果表明:天马产陈皮最强峰位置在1002 cm处,梅江产陈皮最强峰位置在1044 cm处。随着年限增加,双水产陈皮最强相关峰由1094 cm处移至1042 cm处。天马产陈皮最强峰由1002 cm移至1092 cm处。湖北产陈皮最强峰由998 cm移至1092 cm处。直观上,S1 ~S8(广东、广西产陈皮)谱图较为相似,S9、S10、S12、S14 谱图较为相似,S11、S13 谱图较为相似。

图8 陈皮对照药材及14 批样品二维相关红外光谱(1150 ~996 cm-1)Fig 8 2D-IR synchronous correlation spectra of the reference and 14 batches of citri reticulatae pericarpium(1150 ~996 cm-1)
3 讨论
本研究结果表明14 批样品的水分及含量测定均符合药典要求,广陈皮中橙皮苷含量普遍低于陈皮。对于陈皮和广陈皮药材,《中国药典》在含量测定项下的规定也有所不同:陈皮要求橙皮苷含量不低于3.5%,广陈皮要求橙皮苷含量不得少于2.0%,且川陈皮素与橘皮素的总量不得少于0.42%。说明药材的道地性并不通过单一指标性化学成分来评价。仅对现有一种或几种化学成分进行测定,缺乏可信度,所得结果也难以阐明中药道地性。后续将在此基础上进行药效学研究,分析基于中药成分出发的化学评价与药效评价结果间的异同点。
本文对14 批不同产区、不同年限陈皮水分、橙皮苷、川陈皮素、橘皮素含量进行对比分析,并对它们的二维相关红外光谱进行了整体解析。与红外光谱相比,二阶导数光谱可以获得更多的样本信息,能够观察到(1468±2)cm、(1010±5)cm等吸收峰。1700 ~1396 cm波段、1150 ~996 cm波段的二维相关红外光谱结果表明,自动峰的相对强度和最强峰位置可作为陈皮快速鉴别的依据:1700 ~1396 cm波段,2020年四川、2020年湖北产陈皮只含有2个相关峰,广陈皮则含有4 或5 个相关峰。由于相对强度及最强峰位置的不同,2018年浙江与2018年湖南产陈皮的二维谱图在直观上与广陈皮有明显区别,2019年浙江、2018年湖北产陈皮二维图谱与广陈皮较为相似。1150 ~996 cm波段,广陈皮的二维相关谱图形成非常明显的4×4 矩阵。2020年四川、2020年湖北产区陈皮可以被直观有效区分开来,2019年浙江、2018年浙江、2018年湖南、2018年湖北产陈皮则在(1090±1)cm及1044 cm处峰较强,在(1003±1)cm及1060 cm处峰较弱,与广陈皮的二维谱图相比差异较大。
色谱、质谱等鉴别方法具有预处理时间长、过程烦琐、耗时耗材的缺点,建立一种快速、无损、便捷的鉴别手段是亟需解决的问题。已经有许多学者将二维相关红外光谱用于不同产区中药材的鉴别研究,该方法直接对中药粉末进行测定,既可以反映样品整体信息,又具有耗时短、耗材少的优点。以往的研究多数仅对新会陈皮与其他产区陈皮进行鉴别区分,而本文还聚焦了新会内不同产区(双水、天马、梅江等地)陈皮的比较研究,更加细化、具体。二维相关红外光谱法可以为陈皮的品质评价、鉴别、质量控制提供参考。在后续研究中,本课题组将通过细胞筛选平台对上述样品进行功能层面的验证,以期从化学组成和生物学功能多方面探讨样本的差异。
猜你喜欢
家庭医药·快乐养生(2021年10期)2021-10-18
幸福·健康版(2018年3期)2018-03-23
数学学习与研究(2017年20期)2018-01-02
食品与健康(2017年9期)2017-09-13
恋爱婚姻家庭·养生版(2017年2期)2017-02-15
中学数学杂志(初中版)(2016年3期)2016-06-24
Coco薇(2015年5期)2016-03-29
Coco薇(2015年5期)2016-03-29
Coco薇(2015年5期)2016-03-29
Coco薇(2015年5期)2016-03-29
