
苯六甲酸的密度泛函研究
2022-03-25 09:04位艳宾何伟平王德堂
化工技术与开发 2022年3期
位艳宾,何伟平,刘 焕,李 想,王德堂
(1.徐州工业职业技术学院化学工程学院,江苏 徐州 221140;2.江苏省化工新材料工程技术研究开发中心,江苏 徐州 221140)
苯六甲酸常温下为无色斜状晶体,易溶于水和乙醇,存在于多种煤体的氧化热解产物中。王玉高等人[1]采用次氯酸钠和过氧化氢-乙酸酐两种氧化剂,对煤和煤热解半焦粉末进行温和氧化,并对产物进行表征分析。结果表明苯六甲酸存在于芳环缩合程度高的煤体氧化解聚产物中。牛泽世等人[2]对煤沥青分别进行空气预氧化再萃取及先萃取后空气预氧化的处理,对得到的残渣进行碱/O2高温高压氧化,并对比氧化转化率和氧化产物,发现使用先溶剂萃取再空气预氧化的残渣进行氧化,得到的苯六甲酸含量最高。吕璟慧等人[3]采用钌离子催化氧化,对煤热解半焦粉末和石油焦进行逐级氧化解聚,研究结果表明随着氧化级数升高,苯六甲酸的收率逐渐增加。通过定量分析氧化产物中苯六甲酸的含量,可以推测煤中芳环的缩合程度及芳环侧链的连接状态,进而反演煤的分子结构。工业上,将苯六甲酸与浓硫酸、硫酸氢钾混合加热,可制备重要的工业原料均苯四甲酸,杜琨和吴桐等人[4-5]分别以无烟煤、褐煤为原料,尝试制备了均苯四甲酸。另外,苯六甲酸根阴离子作为一种多齿配体,通过羧基上的氧原子与金属离子形成多核链,可以进一步通过氢键连接成一维带状或二维层状的超分子结构[6]。张尽力等人[7]以苯六甲酸为配体合成了Eu(Ⅲ)的二元发光配合物,结果表明配体苯六甲酸的能级与Eu3+离子能级的匹配程度很好,且具有良好的热稳定性。此外,以苯五甲酸与苯六甲酸,或苯六甲酸与苯四甲酸的比值作为指示参数,可以辨识环境中生物炭的来源。常兆峰等人[8]研究发现,采用NaClO氧化处理生物炭,对指示参数无明显影响,表明采用苯多酸分子标志物法识别环境中的生物炭来源,具有高可靠性。鉴于苯六甲酸的潜在来源和重要用途,本文尝试从分子水平上对苯六甲酸的有关性质进行分析和讨论,以期对实验研究提供理论帮助。
1 计算方法
采用B3LYP/6-311+G(d,p)方法对苯六甲酸进行几何结构优化[9],在优化的分子构型基础上,计算了分子的自然键轨道(NBO)及前线轨道(FMO)、均苯四甲酸的制取机理、红外光谱(IR)、核磁共振谱(NMR)和紫外-可见吸收光谱(UV-Vis)。其中,核磁共振谱(NMR)的计算采用GIAO方法,紫外-可见吸收光谱(UV-Vis)的计算采用含时密度泛函(TD-DFT)。
2 结果及讨论
2.1 几何结构
苯六甲酸分子几何结构的优化结果如图1所示,计算所得的键长参数列于表1。与苯甲酸的键长数据[10]比较后可知,苯六甲酸中的苯环与羧基间的C-C键和羧基的O-H键,均比苯甲酸中相应的键长要大,说明苯六甲酸更容易发生脱羧或脱氢反应。表1列出了各化学键的Wiberg键级[11],其中O-H键的键级明显小于其它化学键,苯环与羧基间的C-C键的键级次之,说明在苯六甲酸中,这2类化学键较易断裂。另外,经振动频率分析[12],所得分子的结构无虚频,即证实为稳定分子构型。由图1可知,苯六甲酸存在分子内氢键,从而提高了分子的稳定性;类似地,由于存在多个相邻羧基,电离出1个氢离子后,所形成的酸根可与邻位羧基形成分子内氢键,故有利于羧基上的H+电离,增强了酸性,在一定程度上解释了苯六甲酸为强酸[13-14]。
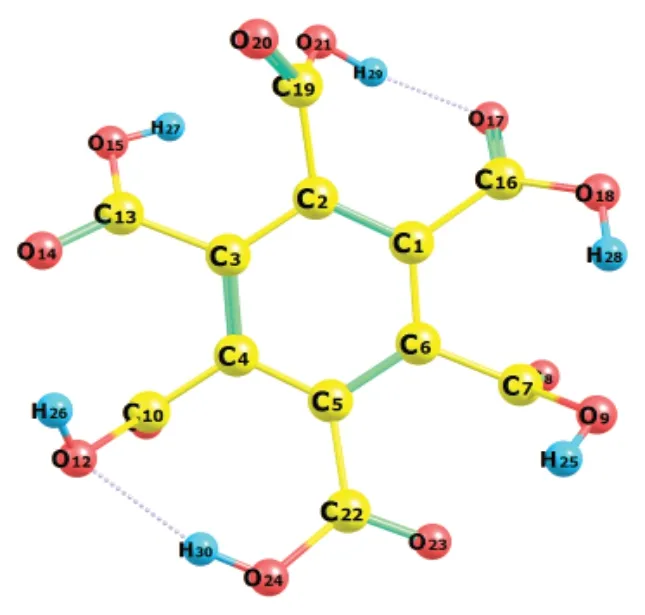
图1 苯六甲酸的分子结构Fig. 1 The molecular structure of benzenehexacarboxylic acid
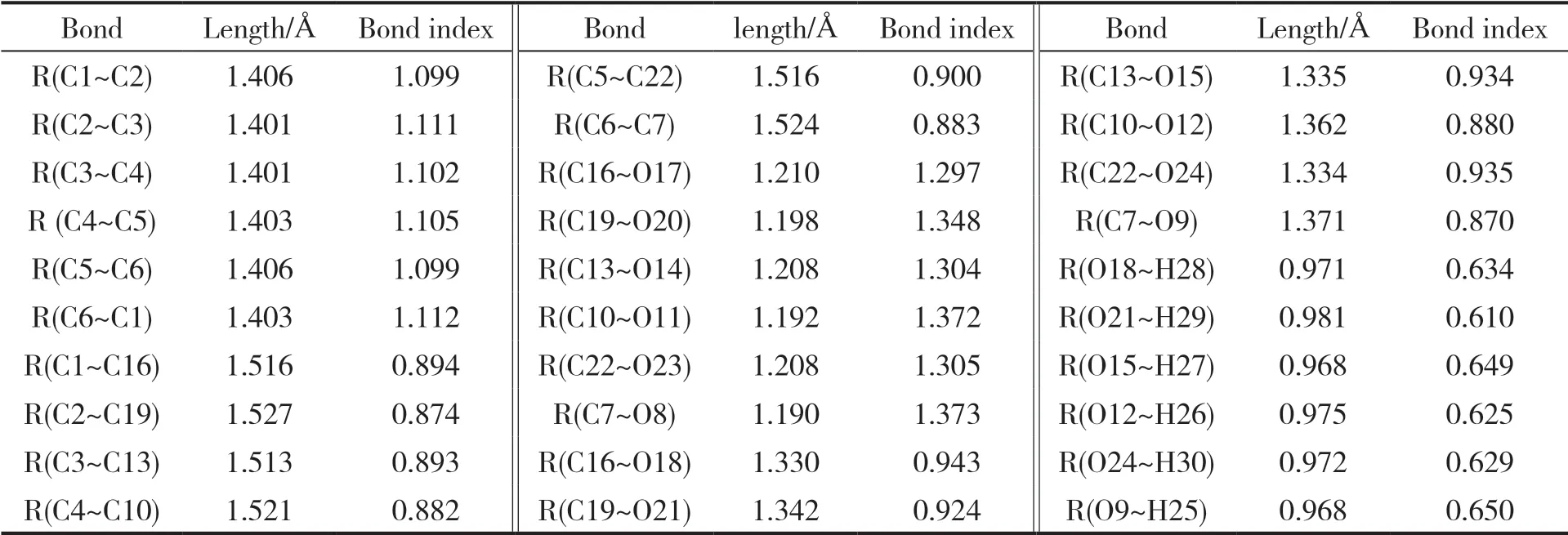
表1 苯六甲酸的键长和键级Table 1 Bond length and Wiberg bond index of benzenehexacarboxylic acid
使用molclus程序搜索团簇构型[15-17],采用半经验方法PM3优化,发现苯六甲酸分子间可通过氢键形成长链结构(图2)。进一步计算二聚苯六甲酸分子的不同构型,发现形成氢键后,体系能量下降了40~60 kJ·mol-1,说明形成的是强氢键[18],在一定程度上说明了苯六甲酸在常温下为固体。
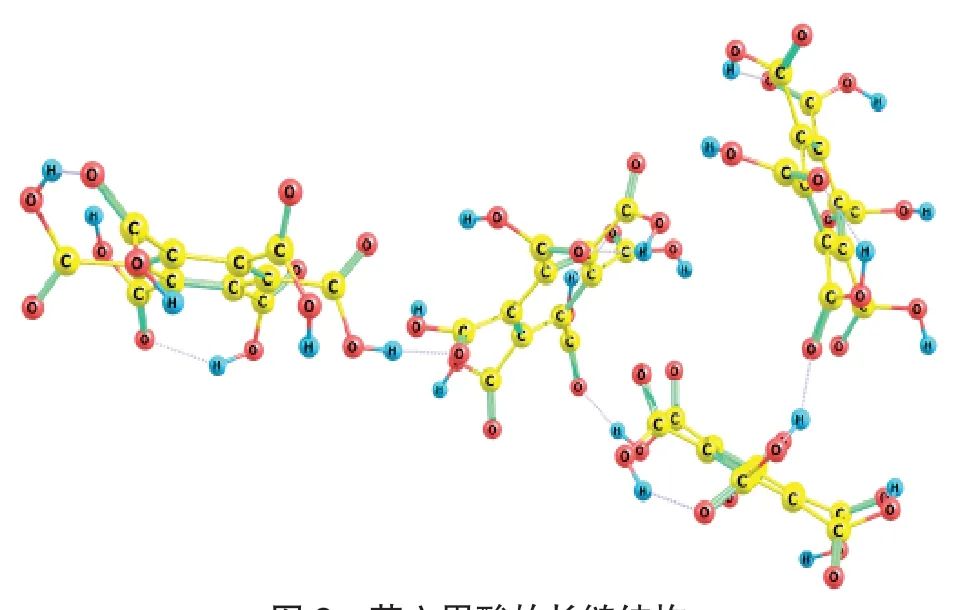
图2 苯六甲酸的长链结构Fig.2 The molecular structure of benzenehexacarboxylic acid
此外,通过计算苯六甲酸与水形成的氢键的构型能量,发现每结合1个水分子,体系能量下降28~33 kJ·mol-1,说明苯六甲酸易溶于水。
2.2 自然键轨道及前线轨道
使用自然键轨道(NBO)方法[19]分析苯六甲酸的电荷性质,计算得到各原子的自然电子组态和自然电荷值[20]见表2。苯环上C1~C6的2s轨道所失去的电荷,比2p、3p轨道得到的电荷少,所以呈现负电荷;C7、C10、C13、C16、C19、C22的2s轨道所失去的电荷,比2p、4s、3d、4p轨道得到的电荷多,因此呈现正电荷。另外,所有H原子的电荷分布几乎没有差异。

表2 苯六甲酸的自然键轨道和电荷Table 2 The NBO and charges of benzenehexacarboxylic acid
前线分子轨道理论认为,化学反应通常发生在前线分子轨道中能级相近和电子密度大的位置上[21],因此前线轨道贡献率[22]大的原子,对分子的反应活性有着重要的影响。
由图3和表3可知,对HOMO贡献率最大的原子为O8,其次为C3、C6、O14,即这些位置在反应中容易与亲电试剂结合。由HOMO→LUMO,分子内电子云的大致流向为O8、C3、C6→C2、C5,可见分子内氢键的形成影响了苯六甲酸的反应位点。
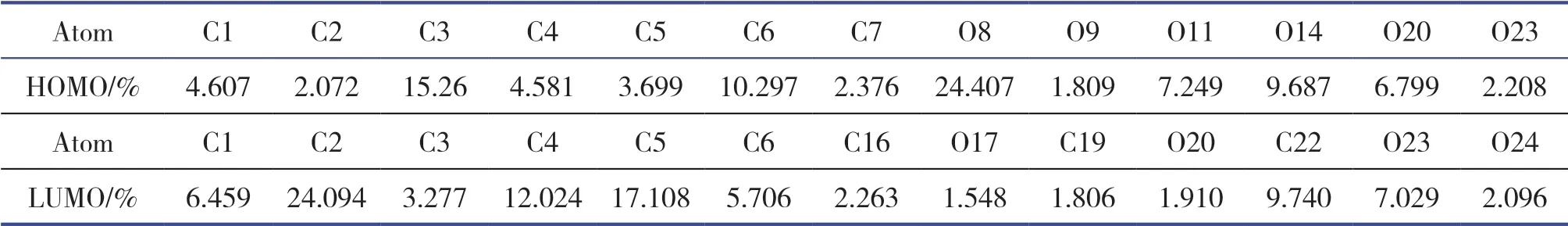
表3 前线轨道组成的分析结果(>1%)Table 3 Frontier orbital composition analysis result(>1%)
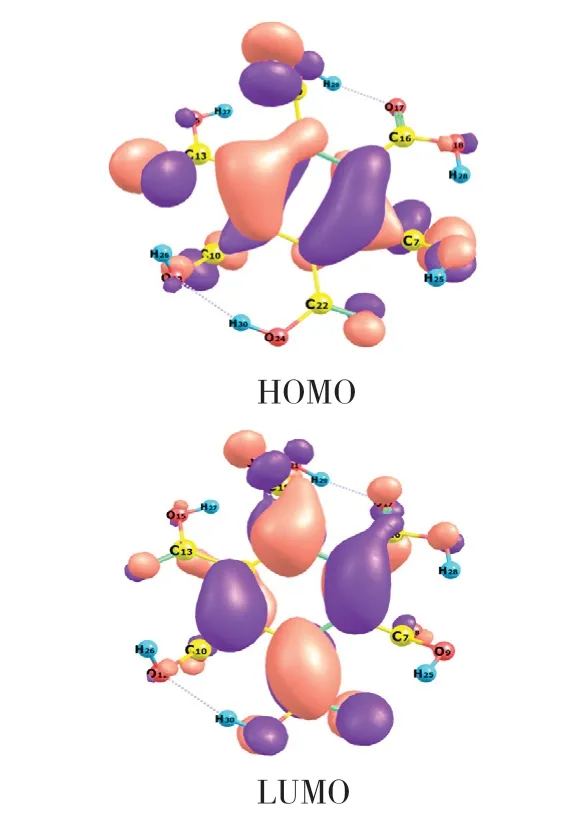
图3 苯六甲酸的前线轨道Fig.3 Frontier molecular orbital densities of benzenehexacarboxylic acid
2.3 均苯四甲酸的制取机理
煤酸(含有苯五甲酸和苯六甲酸)制取均苯四甲酸,是在硫酸氢钾和硫酸介质中、温度高于300℃下进行的,大致经历分子内脱水、脱羧、水解等阶段。根据2.2的结论,苯六甲酸发生分子内脱水反应的位置,应为C2和C3、C5和C6所连接的2对羧基之间,故苯六甲酸的脱水产物如图4所示。
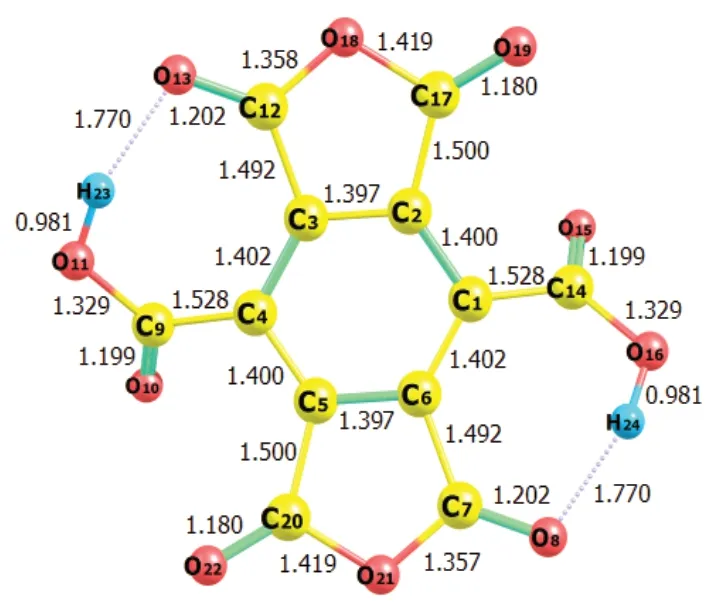
图4 苯羧酸二酐Fig.4 The molecular structure of benzene carboxylic dianhydride
在钾盐的作用下,苯羧酸二酐转化为离子基团,其前线轨道如图5所示。由HOMO→LUMO,分子内电子云的流向为从-COO-流向其他部分,表明在脱羧反应中,-COO-失去电子成为CO2,其他部分获得电子并与质子结合形成均苯四酸二酐,酸性环境则能促进该过程。
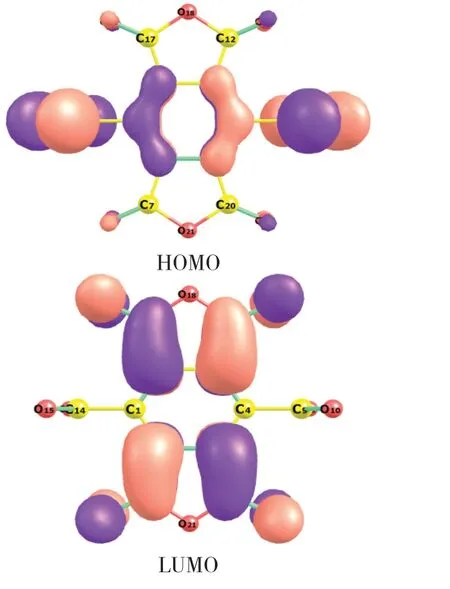
图5 苯羧酸二酐阴离子Fig.5 Frontier molecular orbital densities of benzene carboxylic dianhydride anion
为了描述均苯四甲酸二酐的水解反应活性位点,本文计算了分子的表面静电势[23]。所得静电势的极值点见图6(蓝点代表极小值,红点代表极大值)。由图6可见,均苯四甲酸二酐分子的静电势最小值位于O12附近,次小值位于O10附近,最大值位于C9附近,说明在水解反应中,O12与H结合,C9与O结合,可形成协同的过渡态,最后O10与H结合,形成文献所述的六元环结构[24],即得到产物均苯四甲酸。
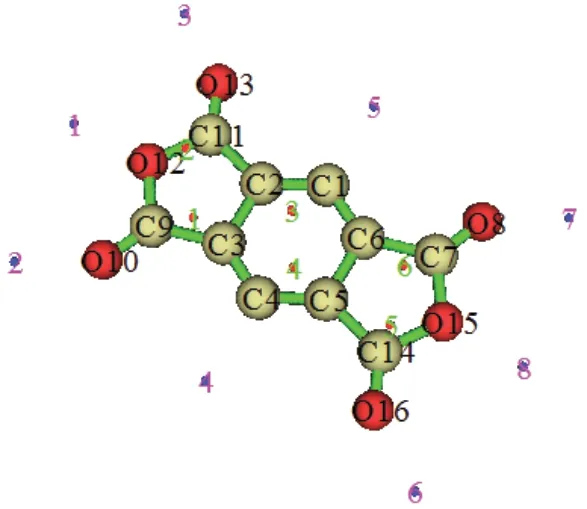
图6 均苯四甲酸二酐的表面静电势Fig.6 The electrostatic potential of pyromellitic dianhydride
2.4 红外光谱
红外光谱具有高度的特征性[25-26],故可用于表征和鉴别各种化学物种。本文红外光谱(IR)的频率校正因子采用0.967[27],红外光谱(IR)的预测结果见图7。观察其振动模式,发现3656cm-1~3410cm-1为O-H的伸缩振动,1798 cm-1~1716 cm-1为C=O的伸缩振动,1520 cm-1~1365 cm-1为苯环骨架的伸缩振动,1333 cm-1~1251 cm-1为C-O的伸缩振动,1213 cm-1~1162 cm-1范围为C-O的伸缩振动和O-H的弯曲振动。对比计算预测的红外光谱与实验测得的红外光谱[28-29]可发现,实测的O-H伸缩振动在3000 cm-1~2500 cm-1范围内,比计算值低得多,这是因为实际上苯六甲酸形成了氢键,对O-H伸缩振动吸收峰的位置和强度均产生了明显的影响,使得伸缩振动频率向低频方向移动。其它振动模式对应的吸收峰位置则非常吻合。
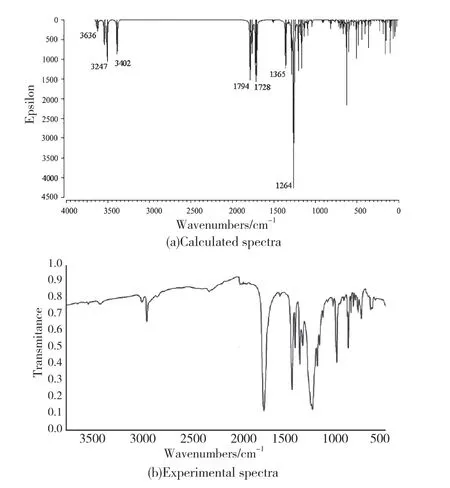
图7 苯六甲酸的红外光谱 (IR)Fig.7 Infrared spectra of benzenehexacarboxylic acid
2.5 核磁共振谱
本文的核磁共振谱以TMS (四甲基硅烷)为内标物,应用规范不变原子轨道GIAO法[30]计算苯六甲酸的核磁共振化学位移d1H-NMR和d13C-NMR,结果如图8所示。
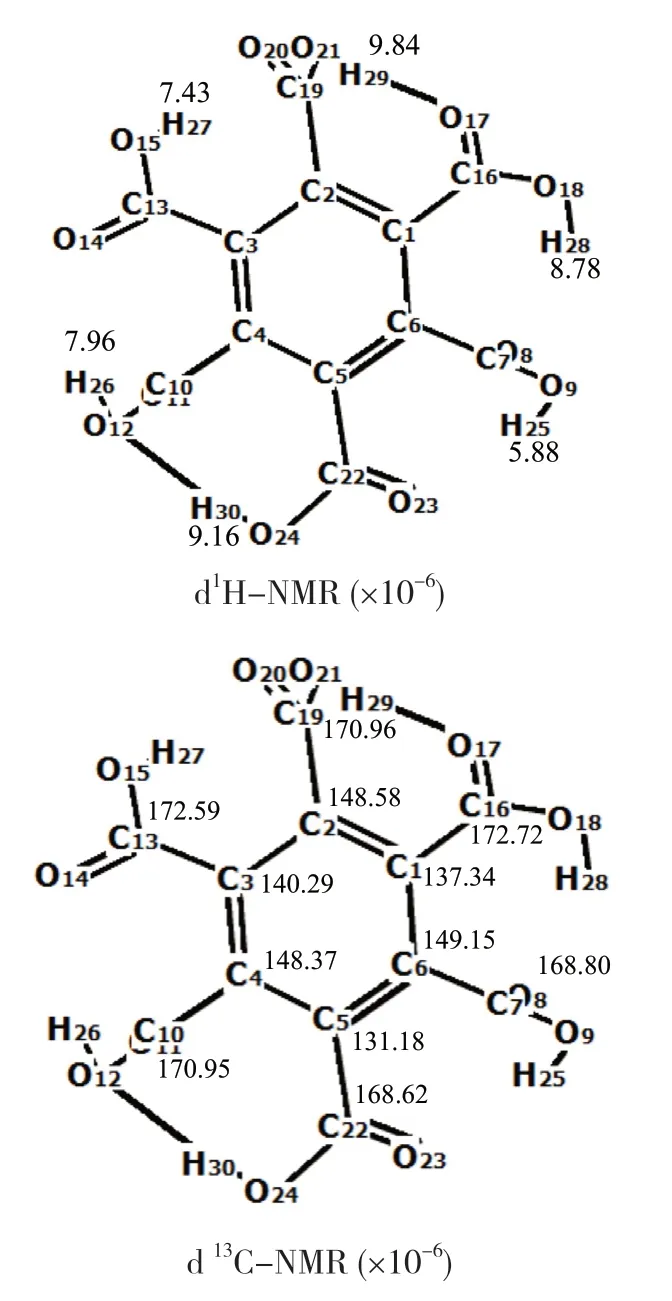
图8 化学位移计算结果Fig.8 Chemical shifts obtained utilizing B3LYP/6-311+G(d,p)
d1H-NMR的计算结果表明,未形成氢键-COOH结构H的d1H值为5.88~8.78,形成氢键-COOH结构H的d1H值为9.16和9.84,证实形成氢键后,H的核外电子云密度降低,化学位移向低场移动。苯环上C的d13C值为131.18~149.15,-COOH结构C的d13C值为168.62~172.72,文献[31]的实测值分别为133.28和166.84,可见预测结果和实测值较为吻合。
2.6 紫外-可见吸收光谱(UV-Vis)
本文计算的苯六甲酸的紫外-可见吸收光谱(UV-Vis)如图9所示。吸收峰波长为269.66 nm,处于紫外区。经计算,得到吸收峰的成分分别为:S0→S6 5.3%、S0→S8 10.4%、S0→S10 9.7%、S0→S13 23.5%、S0→S14 18.3%、S0→S17 21.8%、S0→S20 11.0%,可见S0→S13和S0→S17对吸收峰的贡献最大。
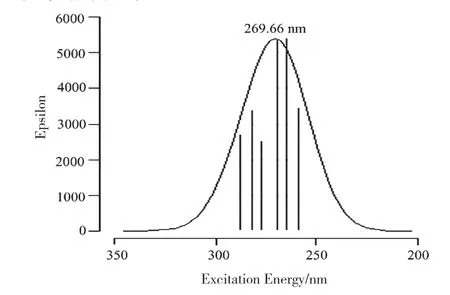
图9 紫外-可见吸收光谱(UV-Vis)Fig.9 Ultraviolet-visible spectrum of benzenehexacarboxylic acid
苯六甲酸基态到各激发态(S0→S13、S0→S17)的垂直跃迁能和振子强度即电子光谱,其主要吸收峰的跃迁波长、振子强度、相应的跃迁方式、跃迁能量等列于表4。可见在S0→S13中,轨道跃迁方式85→88 (LUMO)和87→89(HOMO)对其贡献值最大;S0→S17中,轨道跃迁方式85→88(LUMO)和83→88 (LUMO) 对其贡献值最大。
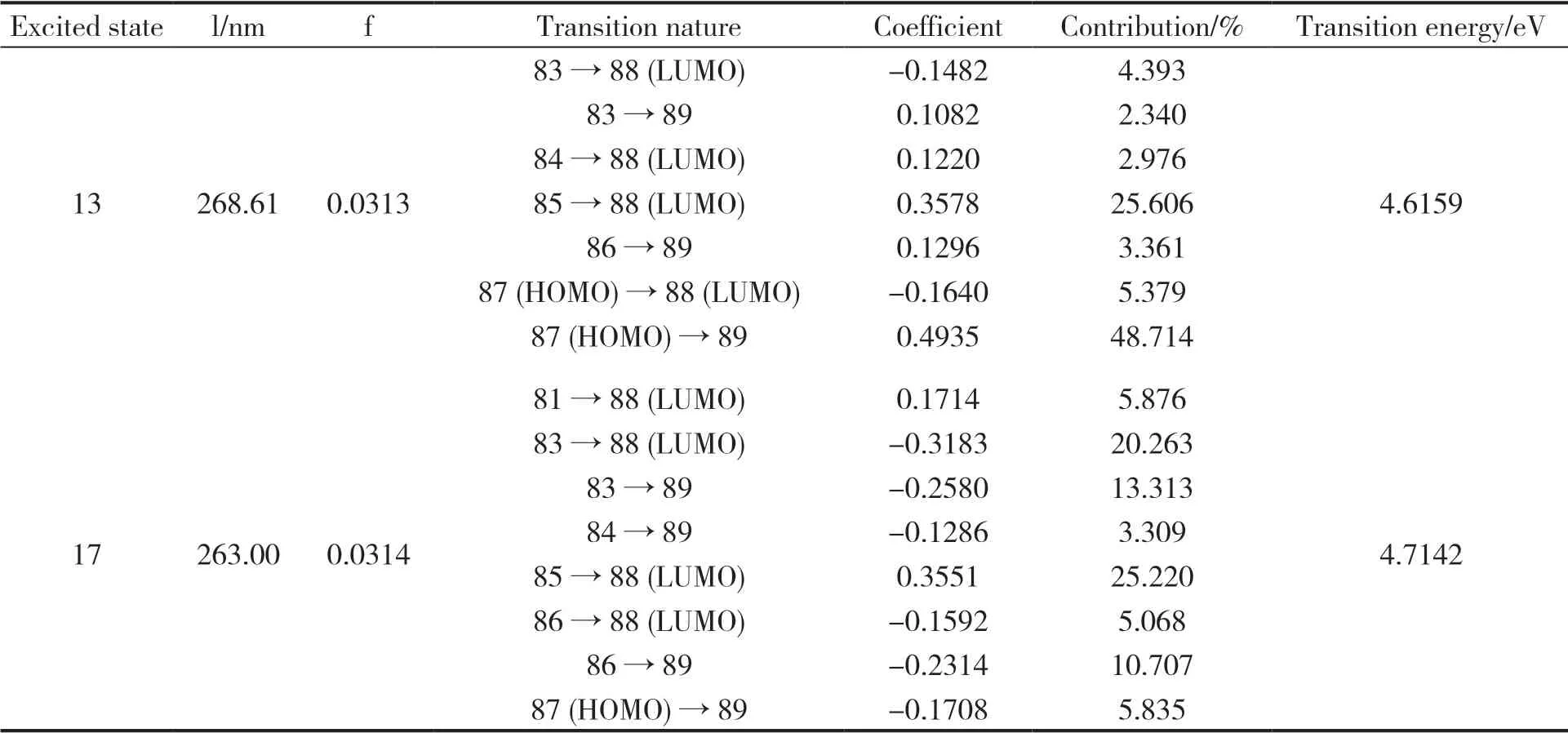
表4 苯六甲酸的电子光谱Table 4 Electronic spectra of benzenehexacarboxylic acid
3 结论
利用密度泛函方法B3LYP/6-311+G(d,p)对苯六甲酸进行了理论计算,得出了苯六甲酸分子的自然键轨道(NBO)、前线轨道(FMO)、均苯四甲酸的制取机理、红外光谱(IR)、核磁共振谱(NMR)和紫外-可见吸收光谱(UV-Vis)。研究结果表明:
1)苯六甲酸分子内部和分子之间存在氢键,从而改变了分子的物化性质。
2)根据自然键轨道及前线轨道,分析原子的电荷分布及分子反应位点,对制取均苯四甲酸的机理进行探讨,结果表明相关理论能较好地解释实验结果。
3)对红外光谱和核磁共振谱进行分析,发现计算得到的数据能较好地解析实验得到的图谱数据。
4)根据紫外-可见吸收光谱(UV-Vis)计算结果,讨论了轨道跃迁方式,揭示了谱图数据的内在形成机理。
猜你喜欢
橡塑技术与装备(2022年8期)2022-12-17
河北师范大学学报(自然科学版)(2022年5期)2022-09-20
中国农业科学(2022年17期)2022-09-19
昆钢科技(2022年2期)2022-07-08
昆钢科技(2021年4期)2021-11-06
波谱学杂志(2021年3期)2021-09-07
——以高中化学“氢键”的教学为例
教学月刊(中学版)(2020年13期)2020-12-29
安徽化工(2018年3期)2018-07-04
分析化学(2018年12期)2018-01-22
山东工业技术(2016年15期)2016-12-01
