
A 型流感病毒非结构蛋白NS1 主要功能的研究进展
2022-03-22 07:38余树芳魏凡华
中国预防兽医学报 2022年1期
余树芳,魏凡华
(宁夏大学农学院,宁夏 银川 750021)
A 型流感病毒(Influenza A virus,IAV)是一种具有包膜的、基因组分节段的负链RNA 病毒,基因组大小约为14 kb。IAV 基因组由8 个RNA 节段组成,大小从890 个碱基到2 341 个碱基不等,可编码最多14 种蛋白:血凝素(HA)、神经氨酸酶(NA)、聚合酶蛋白(PA、PB1 和PB2)、核蛋白(NP)、基质蛋白(M1)、离子通道蛋白(M2)和非结构蛋白(NS1、NS2、PA-X 和PB2-F2)[1]。IAV 具有季节性、地方流行性,是周期性不可预测的全球大流行流感的罪魁祸首。在人类历史上可以追踪到至少5 次流感病毒大流行,分别是1918 年H1N1 西班牙流感大流行、1957 年H2N2 亚洲流感大流行、1968 年H3N2 香港流感大流行、1977 年H1N1 俄罗斯流感和2009 年H1N1流感大流行[2]。当前,2009 年A(H1N1)/pdm2009、H3N2 以及B 型流感病毒是当前主要流行的季节性流感病毒亚型。值得注意的是,多种新型禽流感和猪流感病毒的出现及其在世界范围内的流行,导致人感染禽或猪流感病毒的风险增加,加剧了流感病毒的公共卫生学危害[3]。
流感病毒NS1 蛋白是由流感病毒基因组第8 个节段所编码,大小为202~237 个氨基酸,分子量约28 ku[4]。流感病毒感染宿主细胞中的NS1 蛋白是最先表达的病毒蛋白之一,而且在细胞质和细胞核中的分布具有毒株和亚型特异性[5]。NS1 蛋白尽管在受感染的细胞中大量产生,但它却是一种没有被包装到病毒颗粒中的病毒蛋白。NS1 蛋白由4 个功能结构域组成:N 末端RNA 结合域(RBD,aa1~aa73)、连接区(LR,aa74~aa88)、C 末端效应结构域(ED,aa85~aa202)和C-末端“尾”(CTT,aa207~aa237)[6]。RBD 中的多种碱性氨基酸(如35R、38R、41K 和46R)对于RNA 结合活性和抑制双链RNA 依赖性蛋白激酶R(Protein kinase R,PKR)的激活是至关重要的[7]。ED通过靶向多种宿主因子可以抑制抗病毒反应以及促进病毒复制,如PKR、裂解及腺苷酸化特异因子30(Cleavage and polyadenylation specificity factor 30,CPSF30)和磷脂酰肌醇-3 激酶(Phosphatidylinositol 3-kinase,PI3K)的调节性亚基P85β。ED 结构域的W187残基介导了NS1 蛋白二聚体的形成,W187R 突变减少了NS1 蛋白二聚体的形成,导致病毒毒力的下降[8]。此外,E186、D189和V194残基在NS1 与CPSF30 的结合中起着重要作用,这些位点的氨基酸变异削弱了NS1 与CPSF30 的结合,导致NS1 蛋白的宿主基因关闭功能受损[9]。大量的研究已经证明,NS1 蛋白功能的发挥依赖于它参与的蛋白质-蛋白质和蛋白质-RNA 相互作用的能力[10]。截至目前,已经鉴定出了20多个与NS1蛋白发生相互作用的宿主因子(表1)[10]。总体而言,NS1 蛋白作为一种具有多效性的蛋白质能与多种宿主因子相互作用并通过干扰宿主细胞正常的抗病毒反应,最终促进病毒自身的感染和复制。本文主要从以下三个方面综述NS1 蛋白生物学功能的研究进展:(I)拮抗宿主的抗病毒天然免疫应答;(II)参与细胞自噬过程;(III)介导宿主细胞的凋亡。

表1 与NS1蛋白相互作用的宿主蛋白
1 NS1 蛋白拮抗宿主的抗病毒天然免疫应答
1.1 NS1 蛋白调控IFN-I 信号通路的关键信号分子IAV 感染会触发宿主的天然免疫反应,这是宿主防御的第一道防线。天然免疫系统使用不同的模式识别受体(Pattern recognition receptor,PRR)[包括Toll 样受体(Toll-like receptor,TLR)和维甲酸诱导基因I 样受体(RIG-I-like receptor,RLR)等]来识别侵入的病毒成分。病毒RNA 与PRR 的结合诱导了RIG-I 的构象变化以及K63 连接的RIG-I 的泛素化和寡聚化[11]。寡聚化的RIG-I 与线粒体抗病毒信号蛋白(Mitochondrial antiviral signaling protein,MAVS)结合,进而导致MAVS 的寡聚并募集TBK1 和IKKα/β。随后,TBK1 使干扰素调节因子(Interferon regulatory factors,IRFs)磷酸化从而导致IRFs 的核转位。同时,IKK 介导IκB 的磷酸化和降解能够导致NF-κB的释放。IRFs 和NF-κB 形成活性转录复合物,激活I 型干扰素(IFN-I)的表达(图1)。
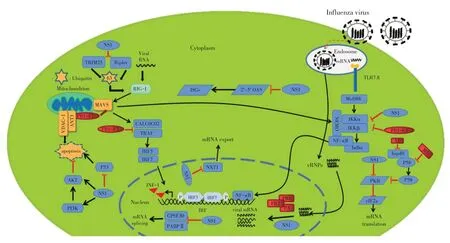
图1 流感病毒激活I型干扰素的示意图
IAV 感染会导致IFN-I 表达上调。IFN-I 能与受感染细胞或邻近细胞中的IFN-α/β 受体结合,以自分泌、旁分泌等刺激抗病毒反应。IFN-I 与受体的相互作用导致Janus 家族蛋白激酶(JAKs)Tyk2 和Jak1 的磷酸化和激活,导致信号分子的募集和激活[如信号转换器和转录激活因子1(Signal transducer and activator of transcription 1,STAT1)和STAT2][12]。激活后,STAT1 和STAT2 与IRF9 形成IFN 刺激基因因子3(Interferon-stimulated gene factor 3,ISGF3),ISGF3 移位到细胞核中可诱导干扰素刺激基因(Interferon stimulated genes,ISGs)的表达[12]。ISGs 产物[如粘病毒抗性蛋白(Myxovirus resistance,Mx)、干扰素诱导的跨膜蛋白(Interferon-induced transmembrane proteins,IFITM)、蛋白激酶R(PKR)和2'-5'合成酶(2'-5'-oligoadenylate synthetase,OAS/RNAse L)等]可诱导宿主细胞的抗病毒状态,这在一定程度上减少了病毒从最初感染部位的继续传播。
NS1 蛋白在感染早期主要定位于细胞核,随后输出到细胞质,是流感病毒编码的最重要的干扰素拮抗蛋白。NS1 蛋白是一种多功能毒力因子,能够与IFN-I 通路中的不同组分相互作用从而抑制宿主抗病毒应答反应[13]。NS1 蛋白通过其位于前73 个氨基酸末端的RNA 结合域与病毒RNA 结合进而阻止其被TLRs 和视黄酸诱导基因I(RIG-I)识别。此外,NS1 蛋白能与宿主细胞蛋白(RIG-I、PKR 和OAS 等)竞争性结合病毒RNA并降低其活性。例如,NS1通过与dsRNA 结合来抑制OAS 的激活[14];NS1 还可以通过结合和抑制TRIM25的活性来抑制RIG-I的激活。
RIG-I 能够识别并结合流感病毒RNA 中的特定结构/序列(例如双链区的5'三磷酸末端)引发其构象变化,暴露其N-端CARD 结构域[15]。暴露的CARD域通过E3 连接酶(如TRIM25 和Riplet)的泛素化作用来激活CARD 结构域,这一过程导致RIG-I 与MAVS的相互作用并激活了IFN-I 的转录。NS1 蛋白可以与TRIM25 结合,从而抑制RIG-I CARD 结构域和IRF3 的激活。也有研究发现,流感病毒NS1 蛋白结合TRIM25 和Riplet 泛素E3 连接酶以抑制RIG-I 泛素化和IFN-I 的产生过程具有宿主特异性[15]。NS1 蛋白还可以诱导去泛素酶OTUB1 的蛋白酶体降解,通过抑制K48 多泛素水解和与UBCH5c 形成E2 抑制复合物的双重机制拮抗RIG-I 抗病毒反应[16]。此外,NS1蛋白通过与p85β 亚基相互作用激活PI3K 通路来增强IRF3的活性从而增加IFN-I和促炎性细胞因子的产生[17]。NS1蛋白还可以通过靶向IKKβ来抑制NF-κB通路的激活以及在细胞核内削弱IKK 对组蛋白3(H3)的磷酸化,进而阻止抗病毒基因的表达[18]。
1.2 NS1 蛋白抑制IFN-I 前体mRNA(pre-mRNA)的加工过程除了抑制天然免疫信号通路外,NS1还通过阻断宿主的mRNA 翻译(例如IFN-I 和ISGs等)抑制宿主基因的表达。NS1 蛋白通过结合两种细胞蛋白:CPSF30 和聚腺苷酸结合蛋白II(PABII),提供了阻断3'末端细胞或病毒前体mRNAs 加工的关键功能。由于NS1 蛋白对CPSF30 的隔离,未加工的细胞前体mRNAs 积累在细胞核中,致细胞质中细胞mRNA 的产生受到抑制[19]。研究发现,不同病毒株结合CPSF30 的能力是不同的。H3N2 和H2N2 病毒的NS1 蛋白能够有效结合CPSF30 并抑制IFN-I 基因pre-mRNA 的加工,然而,H1N1亚型PR8病毒的NS1不能与CPSF30相互作用,而且H5N1病毒NS1蛋白的L103F和I106N突变增加了NS1与CPSF30的相互作用,从而增强了H5N1病毒的致病性[20]。此外,NS1蛋白可以与真核翻译起始因子eIF4B 相互作用,参与宿主基因mRNA的翻译起始。例如,NS1诱导eIF4B蛋白的降解导致了IFN 诱导的跨膜蛋白3(Interferon-induced transmembrane protein 3,IFITM3)的表达减少,进而增加流感病毒的复制水平[19]。总之,NS1 蛋白对宿主基因表达的广泛抑制,不仅抑制了IFN-I 的高效表达也阻止了其他抗病毒基因的表达。
1.3 NS1 蛋白调控炎性小体引发的炎性反应流感病毒感染可激活NLRP3 炎性小体并产生IL-1β,进而促进宿主的天然免疫反应。NLRP3的激活会形成一种由寡聚NLRP3、ASC 和Caspase-1 组成的细胞溶质多蛋白复合物。Toll 样受体或RIG-I 样受体识别病毒RNA,导致pro-IL-1β 和NLRP3 的表达[21]。当NLRP3炎性小体被激活时,会引起ASC 复合物的形成和Caspase-1的激活,导致IL-1β的成熟。
研究发现,NS1 蛋白与NLRP3 的相互作用可以抑制小鼠巨噬细胞系和人THP-1细胞中炎性小体的活化,降低IL-1β的产生[22]。Park等发现携带NS1 C末端缺失的pH1N1/09病毒比野生型病毒诱导的IL-1β水平显著升高,而且仅C 端aa100~aa219 的表达就可以导致IL-1β 的分泌水平降低,这表明NS1 蛋白的C 末端对炎性小体的激活具有抑制作用[22]。进一步研究发现,NS1 蛋白的RNA 结合结构域(碱性残基aa38和aa41)和TRIM25 结合结构域(酸性残基aa96 和aa97)是抑制NLRP3 炎性小体激活所必需的。因此,NS1 蛋白具有抑制NLRP3 炎性小体激活的功能,有助于病毒逃逸宿主的免疫防御。值得注意的是,流感病毒的其他病毒蛋白也可调节NLRP3 炎性小体的激活。例如M2 蛋白和PB1-F2 蛋白均能刺激氧化的DNA 释放从而触发NLRP3 炎性小体的激活,但是这些病毒蛋白触发NLRP3 炎性小体激活的确切机制仍不清楚[21]。因此更好地了解病毒蛋白在调节体内炎症反应和流感病毒感染发病机制中的作用,将有助于开发更为有效的干预措施来治疗与流感病毒相关的疾病。
2 NS1 蛋白调控宿主细胞的凋亡
细胞凋亡,或称程序性细胞死亡,是一个复杂的生物学过程。细胞凋亡主要通过外在凋亡通路(线粒体凋亡通路)和内在凋亡通路(死亡受体凋亡通路)来激活。细胞凋亡是宿主细胞抵御流感病毒感染的一种防御机制,能够抑制病毒的复制和传播[23]。然而,为了更好的促进病毒复制,流感病毒也进化出多种策略来调节宿主细胞的凋亡过程。
多项研究表明流感病毒感染可诱导多种宿主细胞的凋亡。流感病毒在感染初期主要通过上调抗凋亡的磷脂酰肌醇-3-激酶-蛋白激酶B(PI3K/Akt)途径抑制细胞凋亡,在感染后期则通过抑制PI3KAKT 途径以及上调促凋亡的p53 途径进而抑制细胞凋亡[24]。研究表明来自不同流感病毒株的病毒蛋白调节宿主细胞的凋亡过程不同,有的具有促凋亡作用,有的则发挥抗凋亡功能。非结构蛋白NS1 在流感病毒感染诱导宿主细胞凋亡过程中发挥双重功能。早期研究表明,流感病毒NS1 蛋白可以抑制宿主细胞的凋亡过程,而缺失NS1 基因的重组流感病毒诱导宿主细胞凋亡的能力强于野生型病毒[24]。NS1 蛋白抑制宿主细胞凋亡的原因可能与NS1 抑制IFN-I 的产生有关,因为IFN-I 可以诱导细胞凋亡的发生。此外,NS1 蛋白也可以与PI3K 的P85β 调节亚型的SH2结构域或者Crk 或CrkL 的N 末端SH3 结构域结合,激活PI3K/Akt 通路从而延迟流感病毒诱导的细胞凋亡过程,这为病毒复制提供足够的时间[24]。NS1 蛋白诱导宿主细胞凋亡的能力也依赖于流感病毒其他病毒蛋白或宿主因子的参与。例如,宿主因子p53 在介导流感病毒感染引发的细胞凋亡中起着重要作用,NS1 能够与p53结合并抑制p53 介导的宿主细胞凋亡过程,这表明p53 是流感病毒靶向调控感染宿主细胞凋亡的分子之一。综上所述,NS1 蛋白在细胞凋亡中起重要作用,因此靶向定位病毒生命周期不同阶段的NS1 蛋白为治疗IAV 的感染提供了新思路。
3 NS1 蛋白影响细胞自噬
自噬是一种高度保守的细胞内溶酶体依赖的自我消化过程。多种信号参与自噬的诱导,包括内质网应激、氧化应激和免疫信号激活。自噬是消除侵入病原体免疫系统的效应器,并且也参与了病原体侵入的免疫识别。自噬过程可分为两个阶段:自噬体形成的初始阶段和自噬小体与溶酶体的融合并降解自噬体内容物的终末效应阶段(成熟)。其中磷脂酰肌醇-3-激酶III 类(Phosphatidylinositol-3-kinase class III,PI3K-III)、Beclin(Atg6)和LC3(Atg8)是参与自噬的重要因子[25]。
IAV 感染可诱导多种哺乳动物细胞系中自噬体的积聚(如感染IAV 可诱导自噬体在小鼠和人肺组织中的积聚),这说明自噬参与了流感病毒的复制过程。然而,自噬在IAV 复制过程中所起的作用是有争议的,这与细胞类型和病毒株有关[26]。另外,自噬也是宿主细胞抑制流感病毒复制的重要天然免疫机制。流感病毒感染可以诱导自噬体形成,反之诱导流感病毒感染细胞中自噬体的降解过程有助于限制病毒复制。这是由于阻止自噬体的降解过程将导致宿主细胞内自噬体和LC3-II 的积聚以及病毒蛋白抗原识别的逃逸[27]。目前认为,流感病毒的M2、NP、HA 和NS1 蛋白参与了自噬过程。流感病毒的NP 和M2 蛋白均可通过AKT-mTOR 信号通路诱导自噬[26]。H5 和H7 亚型流感病毒HA 蛋白的切割产物可以上调LC3-II 蛋白的水平,说明HA 蛋白可以激活自噬过程[25]。研究发现,NS1 也可以诱导自噬过程,这主要依赖于增加M2 和HA 的合成,从而间接地诱导细胞的自噬过程[25]。
不同亚型流感病毒对自噬具有不同的影响,例如H5N1 亚型流感病毒可以诱导完全和功能性自噬,而H9N2 和H1N1 病毒不能诱导功能性自噬。研究发现,H5N1 病毒通过抑制mTOR 信号通路来抑制细胞的自噬过程,这一过程依赖于肿瘤抑制蛋白2(Tumor suppressor protein 2,TSC2)的调节表达[28]。总之,更好地了解IAV 对自噬的调节过程以及参与自噬调节的各种病毒蛋白的作用,对开发新的抗病毒药物具有重要的理论意义。例如,PI3K 参与自噬膜的形成,而PI3K 抑制剂可以明显降低流感病毒感染早期的病毒滴度和蛋白合成[29]。
4 小结与展望
流感病毒是具有强传染性的人畜共患病的病原,每年造成人和动物的大量死亡,严重威胁了公共卫生安全。禽流感和猪流感的出现,对我国养殖业造成了巨大的经济损失。当流感病毒感染宿主呼吸道细胞时,NS1 对宿主细胞天然免疫应答的抑制作用决定了病毒的致病性。为了进一步解析NS1 蛋白在流感病毒感染中的功能,将来的研究方向应着眼于通过多种分子生物学技术和蛋白组学方法解析NS1 在抑制宿主抗病毒天然免疫应答、诱导宿主细胞凋亡和调节细胞自噬中的详细机制,同时研究NS1 蛋白的其他生物学功能,从而为流感的防控提供一定的理论基础。
猜你喜欢
中国预防兽医学报(2022年6期)2022-12-29
中西医结合心脑血管病杂志(2022年19期)2022-11-19
中国临床解剖学杂志(2022年1期)2022-11-15
检验医学与临床(2022年12期)2022-06-27
中国人兽共患病学报(2022年5期)2022-06-08
临床医学工程(2022年3期)2022-04-20
当代水产(2020年3期)2020-06-15
祝您健康·文摘版(2019年2期)2019-06-11
科学24小时(2019年5期)2019-06-11
家庭百事通·健康一点通(2017年3期)2017-03-22
