
一例Joubert综合征患儿的ARMC9基因新突变
2022-03-21 14:08:20程婷婷吕楠陆超万凯沈玥董纪强朱敏杰罗敏娜尚清高华方
生殖医学杂志 2022年3期
程婷婷,吕楠,陆超,万凯,沈玥,董纪强,朱敏杰,罗敏娜,尚清*,高华方*
(1.国家卫生健康委科学技术研究所,北京 100081;2. 北京协和医学院研究生院,北京 100730;3. 郑州大学附属儿童医院 河南省儿童医院,郑州 450000)
Joubert综合征(Joubert syndrome,JBTS,OMIM#213300)是一种罕见的先天性小脑发育畸形疾病,于1969年由Joubert等[1]学者首次报道。患者的主要临床表现有小脑蚓部发育不全或缺如、脑部磁共振影像(MRI)结果显示“磨牙征”(molar tooth sign,MTS)、肌张力减低、发育迟缓/智力障碍、动眼神经失用、呼吸节律异常等,并可累及肾脏、肝脏、视网膜等器官[2-5]。
JBTS是一种典型的单基因遗传病,主要表现为常染色体隐性遗传模式[2,6]。目前,OMIM(online mendelian inheritance in man,https://www. omim.org)数据库收录与JBTS表型关联的致病基因共39个,这些已知基因的变异能解释约94%患者的患病原因,其中CPLANE1(C5orf42)、CC2D2A、CEP290、AHI1、TMEM67等是突变频率最高的JBTS致病基因[2,7-11]。ARMC9是JBTS的致病基因之一,其变异导致的病例大约占所有JBTS患者的1%[12-13]。
本研究采用全外显子组测序技术对一个JBTS家系的患儿基因组DNA进行检测,筛选出ARMC9基因(NM_025139.4)上的c.485C>G,p. Ser162*和c.1 878+1G>A变异位点,并对患儿及家系成员进行RT-PCR和Sanger测序验证,证实ARMC9的复合杂合变异是该家系的致病原因。
材料与方法
一、实验材料
1.家系来源:该JBTS家系由郑州大学附属儿童医院(河南省儿童医院)康复科采集,包括患儿、同胞姐姐、父亲和母亲。血样以及临床资料的收集均符合知情同意原则。本项研究已通过国家卫生健康委科学技术研究所伦理审查委员会的审查批准(批准文号为201806)。
2.主要试剂:全血基因组DNA提取试剂盒(QIAamp DNA Blood Mini Kit)来自QIAGEN公司(德国),RNA采血管(TempusTMBlood RNA Tube)、RNA提取试剂盒(TempusTMSpin RNA Isolation Kit)、反转录试剂盒(SuperScript IV First-Strand Synthesis System Kit)均来自Thermo Fisher Scientific公司(美国),Easy Taq DNA酶、Fast Pfu DNA酶以及DNA Marker(DL2000)购自全式金生物技术有限公司,PCR扩增测序引物由北京六合华大科技有限公司合成。
二、DNA提取和全外显子组测序
按照QIAamp DNA Blood Mini Kit试剂盒的操作说明从200 μl全血中提取基因组DNA。用NanoDrop2000(Thermo Fisher Scientific,美国)检测DNA浓度。取1 μg DNA用超声仪(Covaris,美国)随机打断成100 bp~200 bp的小片段,使用SureSelect Human All Exon 60M Kit 试剂盒(Agilent,美国)进行建库,利用NovaSeq6000高通量测序仪(Illumina,美国)进行2×150双端测序。
三、测序数据分析
下机数据经质量控制后,使用BWA(Burrows-Wheeler Aligner)软件将数据与GRCh37/hg19基因组进行比对,使用GATK(Genome Analysis Toolkit)和VEP(Ensembl Variant Effect Predictor)[14]进行注释,并参考dbSNP151(http://www.ncbi.nlm.nih.gov/snp)、1000 Genomes Project(http://browser.1000genomes.org)、Genome Aggregation Database(http://gnomad.broadinstitute.org/)及The Human Gene Mutation Database(http://www.hgmd.cf.ac.uk/ac/index.php/)等数据库。变异位点通过下列步骤进行过滤筛选:(1)优先考虑目前已知的JBTS致病基因;(2)保留最小等位基因频率(MAF)<0.01的变异位点;(3)着重关注无义突变、移码突变、经典剪切位点突变等致病性强的变异;(4)按照美国医学遗传学和基因组学学院(The American Collage of Medical Genetics and Genomics,ACMG)指南[15],对筛选后的变异位点进行分级,从而确定候选的致病基因及变异位点。
四、Sanger测序验证
针对ARMC9基因变异位点所在的外显子区域设计上下游引物(引物序列见表1),以患儿及其父母、姐姐的基因组DNA为模板进行PCR扩增,在ABI 3730xl(Thermo Fisher Scientific,美国)上完成Sanger测序。使用Chromas软件查看Sanger测序峰图,SeqMan软件比对Sanger测序结果。
采用反转录PCR(RT-PCR)方法验证c.1 878+1G>A位点在转录水平产生的变化。从全血中提取RNA,逆转录成cDNA,用特异性引物(ARMC9in20F、ARMC9in20R)(表1)进行PCR扩增,并进行Sanger测序。
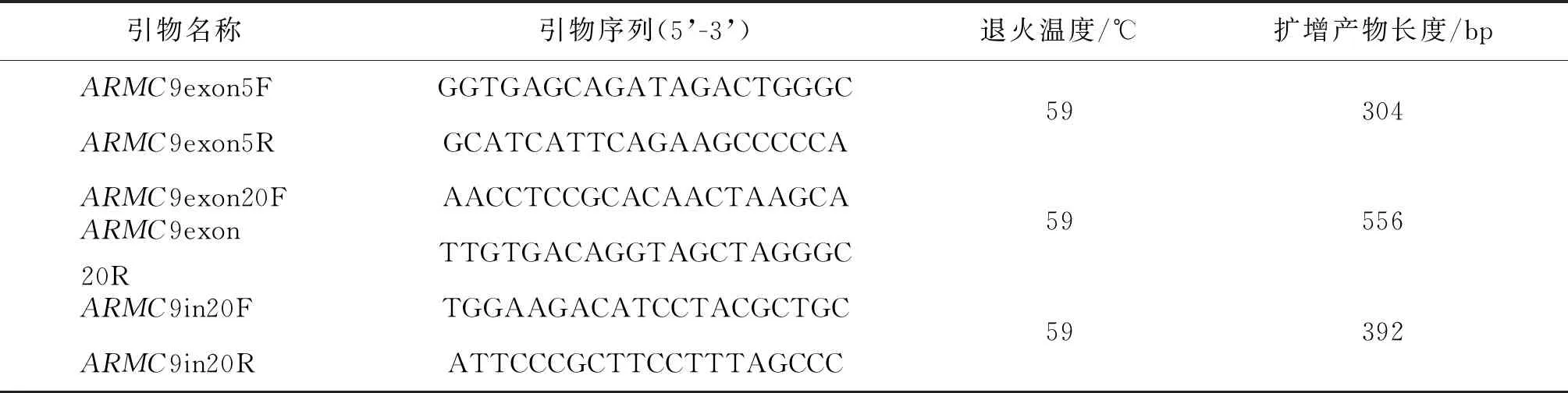
表1 ARMC9基因变异位点扩增引物序列
结 果
一、临床资料
患儿,男,2月10天,以“不会追视、追听”为代主诉至郑州大学附属儿童医院康复科门诊就诊,现病史:不会抬头,对视差,不会追视、追听,易呛奶,睡眠多。出生史:患儿系第3胎第2产,足月顺产,出生体重3.5 kg,出生身长50 cm,出生时有羊水浑浊,无缺氧窒息史,阿氏评分不详。无特殊家族史。查体:头围41.5 cm,右足有六趾畸形,左手小手指根部外侧有肉赘(图1A、B为患儿2岁随访时所拍摄);双眼睑下垂,眼球水平震颤;颈软,心肺腹未见明显异常,膝腱反射可引出,双下肢内收肌肌张力略低,双侧巴氏征阳性,双侧踝阵挛阴性。辅助检查:脑电图未见异常;肝、肾及泌尿系统B超未见异常。血氨基酸及肉碱检测未见异常;视觉诱发电位(flash visual evoked potential,FVEP)异常:双眼P100波潜伏期轻度延长;颅脑MRI异常:双侧额颞部蛛网膜下腔略宽,左侧额部少量硬膜下积液,枕部蛛网膜下腔增宽,小脑蚓部发育欠佳,双侧小脑上脚长、粗(图1C),第四脑室略呈蝙蝠形,脑桥及中脑下部形态欠规则,可见“磨牙征”(图1D),胼胝体部略薄短。临床怀疑Joubert综合征。

A:右足六趾畸形;B:左手小手指根部有肉赘;C:磁共振T1WI矢位示小脑上脚增粗延长;D:磁共振T1WI轴位示“磨牙征”。(箭头所示为病变位置)。
二、基因变异分析
经全外显子组测序,分析发现患儿2号染色体ARMC9基因(NM_025139.4)存在c.485C>G和c.1 878+1G>A复合杂合变异。其中c.485C>G为无义突变,位于第5号外显子,可导致蛋白质翻译提前终止,产生截短蛋白(p.Ser162*);c.1 878+1位于20号外显子的3’端,属于经典剪切位点,c.1 878+1G>A变异为经典剪切位点变异。这两个位点在dbSNP151、1 000 g、gnomAD等公共数据库中未见报道,HGMD数据库未收录,是新的变异位点。根据ACMG指南,均属于致病性变异。ARMC9结构及变异示意图见图2。

A:ARMC9蛋白结构域预测简图,CC:coiled coil结构域;lc:low complexity;B:ARMC9基因变异,黑色字母表示已报道的变异位点,红色字母表示本研究发现的新位点。
三、Sanger测序结果分析
患儿的c.485C>G,p.Ser162*位点来自母亲,c.1 878+1G>A位点来自父亲,其姐携带c.485C>G变异,不携带c.1 878+1G>A变异,符合常染色体隐性遗传的特征(图3)。

A:家系图;B:ARMC9变异位点的Sanger测序峰图(红色箭头示变异所在位置)。
为检测c.1 878+1G>A位点对RNA剪切的影响,分别提取各家系成员的RNA,经反转录得到cDNA后,利用ARMC9in20F、ARMC9in20R引物进行PCR扩增(引物序列见表1)。电泳结果显示父亲(Ⅰ:1)与患儿(Ⅱ:3)存在两个不同长度的扩增条带,母亲(Ⅰ:2)、姐姐(Ⅱ:1)及正常对照(Con)只有单一的扩增条带。Sanger测序结果证实母亲和姐姐只有392 bp的转录本,且序列与正常对照一致,而父亲与患儿除了392 bp的野生型转录本以外,还存在一个287 bp的转录本。经比对分析可知该段序列中缺失了20号外显子的序列。这些结果表明c.1 878+1G>A变异在转录水平可造成外显子跳跃,导致第20号外显子完全缺失(全长105 bp)(图4)。

A:cDNA扩增电泳图;B:正常对照(Con)与患儿(Ⅱ:3)扩增产物的Sanger测序峰图(红色箭头所示为变异起始位置);C:野生型(WT)和c.1 878+1G>A变异(Mut)所导致的转录差异示意图。
讨 论
Joubert综合征是一种罕见的先天性小脑发育畸形疾病,有3个关键诊断标准:(1)颅脑MRI中显示“磨牙征”;(2)婴儿期肌张力减退;(3)发育迟缓/智力障碍,此外还包括异常眼动、呼吸节律异常等症状[2,5-6,16]。该病例MRI显示磨牙征、小脑上脚增粗延长、小脑蚓部发育不良,患儿有肌张力低、发育落后等症状,符合Joubert综合征的诊断标准。此外,该患儿还有多趾、双眼睑下垂、视觉诱发电位异常等表现。全外显子组测序及分析结果表明该患儿为1例ARMC9基因变异所导致的Joubert综合征,其中c.1 878+1G>A变异可导致异常剪切,造成第20号外显子跳跃。
目前对JBTS的临床遗传学研究以国外报道居多,已有至少39个致病基因被发现[11,17-18]。国内的相关研究较少,在已报道的100余例JBTS中仅少数病例具有明确的致病基因和变异位点,这些突变分别位于CPLANE1(C5orf42)、CC2D2A、AHI1、MKS1、NPHP1等14个基因上[19-23]。2021年,Luo等[24]在3个中国人群的JBTS家系中发现了一个新的Joubert综合征致病基因-IFT74(intraflagellartransportprotein74),该基因突变的JBTS患者均表现出“磨牙征”、发育迟缓、多指和轻度唇裂的表型,且所有的4名患者共享c.535C>G变异位点。
本研究中的ARMC9(armadillo repeat containing 9,OMIM#617612)基因,也称KIAA1868,定位于2q37.1,其编码的蛋白质存在两个异构体,长度分别为818个氨基酸和665个氨基酸[12,25]。ARMC9蛋白定位于纤毛的基体,其N端的LisH结构域与微管相连接,在纤毛形成过程中表达上调。ARMC9可以与JBTS相关蛋白CEP104、CSPP1、CEP290、RPGRIP1L、TOGARAM1、CCDC66等发生相互作用。同时,ARMC9/TOGARAM1复合体可调节纤毛微管蛋白的翻译后修饰过程(包括乙酰化和多糖化)、调控纤毛的长度及维持纤毛的稳定性[12,18]。
2017年,ARMC9基因变异首次证实与JBTS相关[12]。Van De Weghe等[12]在11名JBTS患者(其中包括6名男性和5名女性,分别来自8个家庭)中发现了ARMC9基因变异,这些患者具有上睑下垂(7例)、多趾(2例)、视网膜营养不良(2例)等伴发症状。随后,Kar等[13]报道了另一个JBTS家系,此家系中的3名患儿均具有“磨牙征”,同时表现出多指(趾)畸形、智力障碍、上睑下垂的症状。全外显子组测序发现了ARMC9基因的同义突变c.879G>A,p.Thr293Thr。Minigene实验证实该变异在转录水平导致了第9号外显子的跳跃,是该家系的致病原因。2021年初,郑州大学第三附属医院回顾性分析了11例经产前超声及MRI诊断为JBTS胎儿的影像学及遗传学检测结果。其中,有一例具有“磨牙征”、小脑蚓部完全缺失并伴有右足多趾和单脐动脉的胎儿经遗传学检查检出ARMC9基因的3个杂合变异位点,其中c.879+1G>A遗传自母亲,c.1 369C>T和c.348G>A位点遗传自父亲,但此文未对三个变异位点进行详细的解读和致病性分级[26]。
目前HGMD数据库中收录的ARMC9基因变异共13个,包括7个错义突变、2个无义突变、3个剪切位点变异和1个大片段缺失(图2)。这些变异引起的表型多数与JBTS相关,仅有两个位点例外,即c.601G>T(p.Glu201*)和c.443T>C(p.Phe148Ser),它们是在对畸形胎儿的基因筛查中发现的位点[12-13,27-28]。本研究发现的ARMC9基因c.485C>G和c.1 878+1G>A复合杂合变异均未见报道或收录,是新的变异位点。
目前,Joubert综合征的致病机理尚不明确,亦无有效的治疗措施,对高风险家庭进行产前诊断是预防Joubert综合征患儿出生的最有效方法。本项研究所发现的ARMC9基因c.485C>G,p.Ser162*和c.1 878+1G>A变异,明确了该Joubert综合征家系的致病原因,拓宽了ARMC9基因的变异谱,为该家庭的遗传咨询和再生育提供了分子遗传学基础。
猜你喜欢
口腔医学(2021年10期)2021-12-02 02:07:58
中国临床解剖学杂志(2021年2期)2021-04-19 14:52:46
创新作文(小学版)(2019年4期)2019-07-24 09:03:42
哲思2.0(2017年12期)2017-03-13 17:45:04
儿童故事画报·发现号趣味百科(2016年7期)2017-02-08 09:07:45
中华老年口腔医学杂志(2016年5期)2016-03-01 02:24:23
西南军医(2016年2期)2016-01-23 02:14:10
广东海洋大学学报(2015年4期)2016-01-13 08:39:30
听力学及言语疾病杂志(2015年5期)2015-12-24 01:47:04
首都医科大学学报(2015年4期)2015-12-16 13:00:08
