
线粒体动力相关蛋白1基因在胰腺癌顺铂耐药中的作用机制及其对化疗增敏的启示
2022-03-17 07:58郑泽群杨士勇刘子君
中国肿瘤外科杂志 2022年1期
郑泽群, 杨士勇, 刘子君
胰腺癌(pancreatic cancer,PC)指起源于腺管上皮的腺癌,是一种恶性程度较高的消化系统肿瘤[1],具有起病隐匿、恶性程度高、侵袭性强和预后差等特征,5年生存率不足5%[2]。近年来,我国PC的发病率及死亡率均呈逐年递增的趋势。目前,临床上治疗PC的方案主要是采用根治性手术联合辅助性放化疗,以铂类药物为主的化疗是PC一线化疗方案的经典和基础药物,尤其顺铂(cisplatin,DDP)是代表药物之一[3]。Yin等[4]研究证实,DDP治疗早期阶段PC的疗效较好,随着治疗的推进,对DDP等化疗药物产生多药耐药是PC治疗失败的主要原因。因此,研究DDP的耐药分子机制,寻找有效的DDP增敏药物靶点,抑制PC进展、提高PC患者的预后及生存质量成为当前的研究热点。本研究通过探讨线粒体动力相关蛋白1(dynamic related protein,Drp1)基因在胰腺癌DDP耐药中的作用机制,以期对胰腺癌DDP化疗增敏提供新的启示。
1 材料与方法
1.1 PC标本收集 选取2016年1月至2020年12月在南京医科大学附属南京医院行根治性手术治疗的PC患者52例,肿瘤组织均经病理学及细胞学检查确诊,临床资料完整。其中男27例,女25例;年龄35~69(55.3±10.0)岁。肿瘤部位:胰头31例,胰体尾21例;根据第七版TNM分期标准:Ⅰ~Ⅱ期26例,Ⅲ~Ⅳ期26例。按照患者是否对DDP耐药,分为DDP耐药PC组26例,男14例,女12例,年龄37~69(56.3±10.1)岁;DDP非耐药PC组26例,男13例,女13例,年龄35~68(54.1±9.12)岁。留取DDP耐药PC组、DDP非耐药PC组的癌组织和相应癌旁正常组织(距离PC组织边缘2 cm以上,经病理确认无癌细胞浸润的正常组织),置于-80 ℃液氮中保存。本研究经过南京医科大学附属南京医院伦理委员会批准,所有研究对象均签署知情同意书。
1.2 细胞来源和培养 亲本人PC细胞株BXPC-3购自中国科学院典型培养物保藏中心。将BXPC-3细胞接种于含有10%小牛血清的RPMI-1640完全培养基中(含有1%青霉素/链霉素),置于37 ℃、5%CO2饱和湿度培养箱内培养,2~3 d换液1次,于显微镜下观察细胞贴壁情况,待细胞呈对数生长用于后续实验研究。
1.3 DDP耐药PC细胞系BXPC-3/CDDP的建立
选用逐渐递增DDP浓度的方式诱导亲本BXPC-3细胞对DDP耐药。取对数生长期的BXPC-3细胞,接种到含有0.1 μg/ml DDP的RPMI-1640培养基中,培养48 h后弃去培养液,加入RPMI-1640培养基继续培养48 h;传代后,在含有0.2 μg/ml DDP的RPMI-1640培养基中继续培养48 h,弃去培养液,加入RPMI-1640培养基继续培养48 h;后续培养逐渐提高DDP诱导浓度为0.4 μg/ml、0.8 μg/ml、1.6 μg/ml,最终获得一株能耐受1.6 μg/ml DDP的PC细胞系,命名为BXPC-3/CDDP细胞。为了维持PC细胞的DDP耐药表型,将BXPC-3/CDDP细胞培养在含有0.2 μg/ml DDP的RPMI-1640培养基中。
1.4 Drp1质粒的构建、转染及分组 在GenBank中检索人Drp1 cDNA序列(基因编号10059 DNM1L),并据此设计Drp1的引物及探针,其正向序列为:Drp1-HindⅢ, 5′-GAC GAG CTG TAC AAG ATG GAG GCG CTA ATT CCT-3′;反向序列为:Drp1-BamHI, 5′-TTT AAA TTC GAA TTC TCT AGA TCA CCA AAG ATG AGT CTC-3′。pcDNA3.1-Drp1 mut质粒:使用QuickChange试剂盒(安捷伦科技公司)进行Drp1-C644A定点突变,将Drp1-C644A进一步克隆到pcDNA3.1质粒中。使用11.5 μl Lipofectamine 2000(美国英杰生命技术有限公司)与8.25 μl构建的pcDNA3.1-Drp1质粒、pcDNA3.1-Drp1 mut质粒或者pcDNA3.1空载体质粒混合,稀释于750 μl OptiMEM中,室温放置30 min,与PC细胞孵育6 h后,进行后续实验。实验分组如下:①pcDNA3.1组,②pcDNA3.1-Drp1组,③pcDNA3.1-Drp1 mut组。将BXPC-3/CDDP细胞转染相应质粒,培养48 h后,以20 μg/ml DDP作用细胞24 h,实验分组如下:①DDP组,②DDP+pcDNA3.1-Drp1 mut组,③DDP+pcDNA3.1-Drp1组。
1.5 免疫印迹法(Western blotting)检测Drp1蛋白表达水平 采用1 ml蛋白裂解液RIPA将PC组织样本(20 μg)匀浆成糊状,4 ℃条件下以15 000×g离心20 min,收集上层胞浆蛋白。取30 μg蛋白,沸水浴使蛋白变性,用10%~12% SDS-PAGE进行电泳分离,湿转法将凝胶蛋白转移至PVDF膜,5%脱脂牛奶室温封闭2 h;用一抗Drp1(ab184247, 英国艾博康公司)及actin(ab8227, 英国艾博康公司)进行4 ℃孵育过夜;再与HRP标记的二抗室温孵育1 h,ECL发光试剂显影,用Bio-Rad凝胶成像分析仪对印迹条带进行灰度扫描分析,以actin灰度值为内参照计算目的条带蛋白含量。
1.6 应用荧光染料分子探针2,7,-二氯荧光黄双乙酸盐(H2DCFDA)检测PC细胞内活性氧(ROS)含量
将PC细胞密度调至1×106/ml,接种至6孔培养板,转染相应质粒培养48 h,加入H2DCFDA(10 μmol/L),置于37 ℃、5%CO2培养箱中静置30 min,3~5 min混匀1次,使荧光探针与PC细胞充分结合;倒置荧光显微镜下选择激发波长488 nm和发射波长530 nm检测细胞内ROS含量,以相对未转染pcDNA3.1质粒的PC细胞组的荧光强度作为细胞内ROS含量。
1.7 新型荧光探针四氯四乙基苯并咪唑基羰花青碘化物(JC-1)检测线粒体膜电位 采用JC-1标记线粒体,检测PC细胞线粒体膜电位变化;将PC细胞密度调整至1×106/ml,接种于6孔培养板,转染相应质粒培养48 h,加入JC-1染色工作液(10 μmol/L),37 ℃条件下静置20 min;荧光显微镜下选择激发波长485 nm和发射波长530 nm,检测JC-1单体的变化,以相对未转染pcDNA3.1质粒的PC细胞组的荧光强度作为细胞内线粒体膜电位。
1.8 ATP生物发光检测线粒体ATP水平 将PC细胞浓度调整至1×106/ml,接种至6孔培养板,转染相应质粒培养48 h,通过添加0.6 mg/ml寡霉素前后ATP生成量之差换算线粒体ATP的含量(寡霉素具有抑制线粒体ATP合酶的作用)。采用TD-20/20光度计(美国特纳公司)检测ATP含量。
1.9 甲基噻唑基四唑(MTT)比色法测定PC细胞增殖活性 将PC细胞接种至96孔培养板,每孔1 000~10 000个细胞,转染相应质粒,培养48 h,以20 μg/ml DDP作用24 h后,每孔加入0.5%MTT溶液20 μl,继续培养4 h,弃去培养上清。每孔加二甲基亚砜(DMSO)150 μl,震荡10 min,使结晶物充分溶解;酶联免疫检测仪于490 nm波长处测定各孔的光密度(OD)值,以时间为横坐标,OD值为纵坐标,绘制PC细胞的生长曲线。
1.10 Transwell法检测PC细胞迁移能力 采用膜孔径为8 μm的Transwell小室(美国康宁公司)检测PC细胞的迁移情况。BXPC-3/CDDP细胞转染相应质粒,培养48 h后,以20 μg/ml DDP作用24 h,收集细胞,以无血清培养基调整细胞密度为3×106个/ml;取200 μl细胞悬液置于Transwell小室上室,下室加入500 μl含20% FBS的培养基,继续培养48 h后,下室表面以0.1%结晶紫染色,在200倍显微镜下计数贴壁细胞,随机选取3个视野的平均细胞数,以此评估PC细胞的迁移能力。
1.11 流式细胞仪检测PC细胞凋亡情况 BXPC-3/CDDP细胞转染相应质粒,培养48 h后,以20 μg/ml DDP作用24 h,胰酶消化;以结合缓冲液调整细胞密度为1×106个/ml,加入5 μl Annexin V-异硫氰酸荧光素(FITC)和10 μl碘化丙啶(PI)溶液,室温避光孵育30 min;于1 h内用FACSCalibur系统采用流式细胞术分析BXPC-3/CDDP细胞的凋亡率。结果判定:在流式细胞仪散点图中,左下象限FITC-/PI-,为活细胞;右上象限FITC+/PI+,为坏死细胞;右下象限FITC+/PI-,为凋亡细胞。
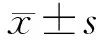
2 结果
2.1 Drp1蛋白在PC组织中的表达 在DDP非耐药PC组,PC组织与其对应的癌旁正常组织的Drp1蛋白表达量比较,差异无统计学意义(P>0.05);而在DDP耐药PC组,PC组织中Drp1蛋白表达量低于其对应的癌旁正常组织,差异有统计学意义(P<0.001),见图1、表1。

R:DDP耐药PC组织;NR:DDP非耐药PC组织;N:PC癌旁正常组织图1 Drp1蛋白在PC组织中的表达
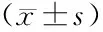
表1 Drp1蛋白在PC组织中的水平比较
2.2 Drp1基因对PC细胞线粒体功能的影响 与pcDNA3.1组比较,pcDNA3.1-Drp1组ROS水平增加,线粒体膜电位及线粒体内ATP含量减少,差异有统计学意义(P<0.05);与pcDNA3.1组比较,pcDNA3.1-Drp1 mut组中ROS水平减少,线粒体膜电位及ATP含量增加,差异有统计学意义(P<0.05);与pcDNA3.1-Drp1组比较,pcDNA3.1-Drp1 mut组ROS水平减少,线粒体膜电位及线粒体内ATP含量增加,差异有统计学意义(P<0.05,见图2、表2)。

表2 Drp1基因对PC细胞线粒体功能的影响比较
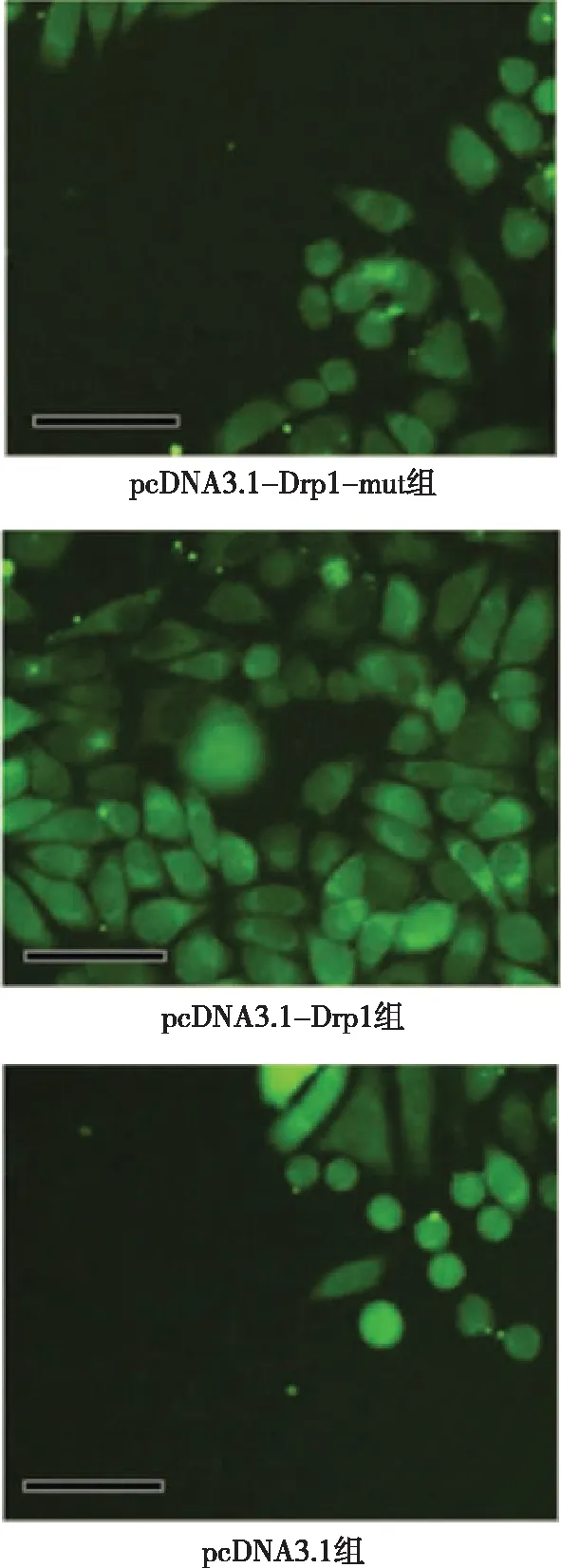
图2 Drp1基因对PC细胞线粒体ROS产生的影响 (×200)
2.3 Drp1基因对DDP耐药PC细胞生物学功能的影响 与DDP组及DDP+pcDNA3.1-Drp1 mut组比较,DDP+pcDNA3.1-Drp1组细胞的增殖活性及迁移数量减少,细胞凋亡率增加,差异有统计学意义(P<0.05);DDP+pcDNA3.1-Drp1 mut组与DDP组细胞的增殖活性、迁移数量及凋亡率比较,差异无统计学意义(P>0.05),见图3、4,表3。

图3 各组BXPC-3/CDDP细胞的迁移情况 (×200)

图4 各组BXPC-3/CDDP细胞的凋亡情况
表3 各组PC细胞的增殖活性、迁移数量及凋亡率比较
3 讨论
PC临床治疗多采用手术为主、放疗和化疗为辅的综合治疗方案,能够缓解临床症状,缩小原发病灶,减缓肿瘤复发[5]。但手术切除术对PC的治疗效果有限,因此有效、敏感化疗药物对于PC治疗显得尤为重要。DDP作为一种无机铂剂,是针对细胞周期的非特异性药物,其对多种肿瘤具有显著的疗效,包括卵巢癌[6]、膀胱癌[7]、恶性淋巴瘤[8]、宫颈癌[9]及结直肠癌等[10]。DDP可通过诱导癌细胞DNA-蛋白、DNA链内或链间交联等方式,抑制癌细胞增殖同时促进癌细胞的凋亡,但癌细胞对DDP诱导的损伤会进行自我切除修复,由此产生类似化疗药物甚至不同机制的其他化疗药物产生的多药耐药现象[11]。尽管DDP对正常细胞具有毒性作用,并且伴随耐药的发生,但DDP依旧是许多实体瘤的一线治疗方案。由于PC恶性程度极高,病情进展迅速,对放化疗敏感度逐步降低,因此,寻找有效治疗策略增加DDP耐药PC治疗增敏靶点对PC患者尤为重要。本研究诱导了一株获得性DDP耐药的PC细胞株BXPC-3/CDDP,以BXPC-3/CDDP细胞为实验模型,探讨PC细胞DDP耐药的机制及DDP增敏的潜在药物作用靶点。
近年来,在临床中发现多种肿瘤细胞对DDP出现耐药现象,其中线粒体动态变化在DDP耐药中发挥至关重要的作用[12]。线粒体作为生物细胞中的一种重要细胞器,一方面作为细胞的动力工厂为细胞提供能量代谢,另一方面参与细胞蛋白质合成、遗传、氧化还原和耐药性的产生。正常情况下,线粒体分裂与融合所形成的动态平衡对于维持线粒体功能至关重要,其参与了肿瘤的发生发展及肿瘤耐药机制的形成[13]。作为一种GTP酶,Drp1基因可以介导线粒体分裂、导致线粒体碎片化,诱导线粒体凋亡的发生。尽管Drp1基因主要定位于细胞质,但应激状态下被招募到线粒体,诱导线粒体分裂和(或)线粒体自噬的发生[14]。研究表明,ROS在缺陷的线粒体中累积,诱导线粒体损伤的发生,进而导致线粒体膜电位上的非特异性孔打开,线粒体中凋亡相关因子释放到细胞质中,由此引发细胞受损甚至凋亡[15]。本研究显示,过表达Drp1基因可以诱导BXPC-3/CDDP细胞线粒体ROS的产生,破坏线粒体膜电位的同时,细胞内ATP含量显著减少,即Drp1基因的过度表达导致BXPC-3/CDDP细胞线粒体功能紊乱,进而引发细胞凋亡的发生。
本研究发现DDP耐药PC组织中的Drp1蛋白表达量显著低于其相应的癌旁组织,即DDP耐药的PC组织中Drp1基因呈现低表达的特征。因此,由Drp1基因诱导的线粒体分裂与融合动态失衡可能与PC细胞DDP耐药的机制息息相关,基于此,以Drp1基因为立足点探索PC细胞DDP耐药具有可行性。本研究为进一步证实Drp1基因在DDP耐药PC中的重要角色,将BXPC-3/CDDP细胞中过表达Drp1基因,观察DDP作用下PC细胞耐药性的改变,结果表明,过表达Drp1基因与DDP联合作用显著抑制细胞的增殖活性和迁移数量,且BXPC-3/CDDP细胞的凋亡也显著增加;然而,增强表达Drp1变异体基因,DDP对BXPC-3/CDDP细胞的凋亡作用显著降低,在凋亡诱导前抑制Drp1活性不仅减少线粒体分离,还能延缓Caspase-3的活化剂细胞死亡过程[16]。
综上所述,本研究结果显示,Drp1基因不仅能够诱导PC细胞线粒体功能紊乱,还能促进BXPC-3/CDDP细胞对DDP的敏感度,改善DDP耐药细胞株的化疗抗性,其可能通过促进细胞中ROS的产生、破坏线粒体膜电位以及抑制细胞内ATP的产生,最终促进DDP诱导的细胞凋亡。本研究进一步证实联合Drp1基因治疗DDP耐药的PC的有效性,有望为针对PC的DDP增敏提供新的思路和依据。
猜你喜欢
南京医科大学学报(自然科学版)(2022年8期)2022-11-22
重庆理工大学学报(自然科学)(2022年9期)2022-10-26
中学生物学(2022年8期)2022-10-13
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中国典型病例大全(2022年7期)2022-04-22
三农资讯半月报(2020年11期)2020-06-21
江苏农业学报(2019年1期)2019-09-10
保健与生活(2019年2期)2019-08-01
中国医药导报(2019年13期)2019-06-20
