
Bacterial community composition in gut content and ambient sediment of two tropical wild sea cucumbers ( Holothuria atra and H. leucospilota)*
2022-03-05 09:36:40FeiGAOYueZHANGPeilinWUMenglingCHENLinwenHEQiangXUAiminWANG
Fei GAO , Yue ZHANG , Peilin WU , Mengling CHEN , Linwen HE , Qiang XU ,**,Aimin WANG
1 State Key Laboratory of Marine Resource Utilization in South China Sea, Hainan University, Haikou 570228, China
2 Ocean College, Hainan University, Haikou 570228, China
Abstract Sea cucumbers play an important role in sediment bioturbation in coral reef and rocky intertidal ecosystems, and bacteria were a commonly-reported component of holothuroid diets. Bacterial community composition in the foregut and hindgut of two common tropical sea cucumbers ( Holothuria atra and H.leucospilota) and the ambient surface sediment were investigated using high throughput sequencing based on 16S rRNA gene analysis. A total of 5 584 operational taxonomic units (OTUs) were obtained from 25 samples based on a 97% threshold, and more than half of the OTUs ( n=3 694, 66.2%) were shared by the gut contents of two species of sea cucumbers and surrounding sediments. Bacterial richness and diversity in sediment samples were signif icantly higher than those in the gut content samples ( P <0.05).Proteobacteria was the predominant phylum in most samples showing 45.69%±8.61%, 70.09%±4.03%,45.88%±5.38%, and 55.19%±0.79% reads in the foregut of H. leucospilota, hindgut of H. leucospilota,hindgut of H. atra, and sediment libraries, respectively, but Bacteroidetes was the predominant phylum with the relative content of 34.98%±5.52% in the foregut of H. atra. Among the dominant genera, reads related to the genera Anderseniella, Ilumatobacter, and Ruegeria were detected in all the gut contents and sediment libraries. A comparison of gut bacteria community between the two species of sea cucumbers suggested that H. atra had stronger feeding preference than H. leucospilota, and the same types of microbes escaped digestion of the two sea cucumber species. Obvious diff erent bacterial community composition in the foreguts of the two species of sea cucumbers and the surrounding sediments might result from the animal’s selective feeding for sediment patches. Bacterial community structure in hindgut contents of H.atra and H. leucospilota both diff ered clearly from adjacent sediments, which indicated feeding activity of deposit-feeding sea cucumbers could change the sedimental bacterial composition. In conclusion, from the perspective of bacteria, sea cucumber H. atra and H. leucospilota had diff erent feeding preferences, yet they could both aff ect bacterial composition in sediments by feeding activity. The motivation for selective feeding and sea cucumber-sediment interaction might be explored in the future.
Keyword: sea cucumber; bacteria; gut; high-throughput sequencing; sediment
1 INTRODUCTION
Sea cucumbers (Echinodermata: Holothuroidea)are important benthic marine invertebrate resources found in all of the world’s major oceans and seas. Up to date, there are 1 750 accepted extant holothuroid species in the world (WoRMS, 2020). Of these, about 66 species of sea cucumber are commonly exploited in multiple locations (Purcell, 2010; Purcell et al.,2012; Conand, 2018). In Asia and the Middle East,some sea cucumbers are considered delicacies, a source of protein, folk medicine, or an important part of income (Bordbar et al., 2011; Purcell et al., 2012;Yang et al., 2015). As large and abundant members of marine benthic communities, their feeding and excretion have crucial eff ects on the physicochemical processes of soft-bottom and reef ecosystem. They can be the host of many species of symbionts and parasites, thus enhancing biodiversity; and they are preyed by many taxa, thereby transferring nutrients from detritus to higher trophic levels (MacTavish et al., 2012; Schneider et al., 2013; Purcell et al., 2016).
Bacteria are vital in coral reef processes, such as organic matter decomposition and nutrient recycling in reef food chains. Because of the high abundance,production, and nutritional values, the bacteria in the sediment are a direct food source or a source that indirectly provides the host with essential nutrients,such as B-complex vitamins for marine detritus feeders(Gerlach, 1978; Deming and Colwell, 1982; Moriarty,1982; Phillips, 1984; Amaro et al., 2009). As typical common detritus feeders, sea cucumbers primarily digest bacteria, decaying seagrass and algae, diatoms,f lagellate, protozoan, foraminiferans, fungi, and other organic matter, which constitute detritus (Yingst, 1976;Moriarty, 1982; Uthicke, 1999; Gao et al., 2010;MacTavish et al., 2012). In particular, bacteria have been a commonly-reported component of holothuroid diets (Moriarty, 1982; Gao et al., 2010; Wen et al.,2016). Feeding activity of sea cucumbers takes a strong part in the bioturbation of marine benthic ecosystems by removing considerable amounts of sediments during gut passage (Bonham and Held, 1963; Mangion et al.,2004), so we supposed sea cucumbers could aff ect the sediment bacterial community structure through their ingestion and digestion processes.
Sea cucumbersHolothurialeucospilotaandH.atraare both exploited as edible sea cucumbers belonging to the genusHolothuria. These sea cucumbers naturally distribute in tropical seas. Their populations are relatively dense and shallow subtidal,commonly found on inner and outer reef f lats, back reefs, and shallow coastal lagoons (Purcell et al.,2016; Conand, 2018). They are known as depositfeeders, gathering organic detritus and sediments from the sediment surface. Nevertheless, for these two sea cucumber species, the researches mainly focus on taxonomic identif ication (Wen et al., 2011),extraction of bioactive materials (Han et al., 2007;Ibrahim, 2012; Esmat et al., 2013; Kitisin et al., 2019;Darya et al., 2020; Zhao et al., 2020), breeding and rearing (Huang et al., 2018), and ecological eff ects on recycling nutrient in reefs (Bonham and Held, 1963;Vidal-Ramire and Dove, 2016; Viyakarn et al., 2020).
However, forH.leucospilotaandH.atra, the gut bacterial community composition and their eff ects on sediment bacterial community by their feeding activity have been surveyed sporadically. The amounts of culturable aerobic bacterial f lora in the digestive tract ofH.atraand the surrounding sediment were investigated, and some isolates were identif ied by 16S ribosomal DNA (16S rDNA) sequence analysis(Ward-Rainey et al., 1996). Zhang et al. (2012) isolated and identif ied 141 bacterial strains under aerobic conditions from the gut ofH.leucospilota, and detected the polysaccharide degradation activities by plate methods using carboxymethyl cellulose sodium salt, xylan, alginate, starch, or agar as the substrate.The amounts of culturable bacteria were measured in the substrate, anterior and posterior gut ofH.scabra,H.atra, andH.leucospilota(Hatmanti and Purwati,2011). Muramic acid, the component of bacterial cell wall peptidoglycan, is often used as an indicator of the content of bacteria in environmental samples. Moriarty(1982) determined the bacterial biomass by muramic acid measurements in the sediments and gut contents ofH.atraon the Great Barrier Reef. The previous research mainly studied the gut microbiota, which could be cultured in sea cucumbers. As most of the bacteria in the nature are non-culturable under laboratory conditions, the characteristics of a tiny proportion of the total bacterial community in the gut of sea cucumbers were revealed. Thus, it is necessary to apply culture-independent methods to improve the understanding of total microbiota in the digestive tract and sea cucumbers’ habitat. High-throughput sequencing techniques have been used to analyze the microbiota in various biomes, such as the gastrointestinal tracts of humans and animals, soils,water, and sediments (Warnecke et al., 2007; McLellan et al., 2010; Hess et al., 2011; Gao et al., 2014a; Jia et al., 2020).
In this study, we examined the bacterial community composition in the habitat surface sediment, the foregut and hindgut contents of two species of common tropical sea cucumbersH.atraandH.leucospilotaby 16S rRNA gene high-throughput sequencing. The objectives of this study were: 1)characterize and compare the bacterial community diversity and abundance in gut contents of the two species of sea cucumbers; 2) compare the bacterial community structures ingested by sea cucumbers (in foregut) with the surrounding sediments so as to investigate the sea cucumbers’ feeding selectivity concerning to sediment bacteria; 3) compare the bacterial community structures in the hindgut of sea cucumbers with the surrounding sediments in order to identify eff ects of the feeding of sea cucumbers on environmental microorganisms.
2 MATERIAL AND METHOD
2.1 Material
Sea cucumbers were collected from a natural distribution area locating in the intertidal zone of Tongguling natural reserve in Wenchang, Hainan Province, China.H.leucospilota(average wet weight of 392.10±71.89 g (n=5)) andH.atra(average wet weight of 131.84±11.98 g (n=5)) individuals were collected on April 20, 2019. Ambient surface sediments (0-1.2 cm,n=5) were collected accordingly from 5 locations around the sampled sea cucumbers using 50-mL syringe samplers (Michio et al., 2003;Gao et al., 2014a). The water temperature and salinity at the sampling site was 28.4 °C and 31. Then, the sea cucumber and sediment samples were stored on ice and transported to the laboratory immediately.
Under sterile conditions, sea cucumbers were washed with sterile seawater, and the scarfskin was sterilized with 75% ethanol to reduce exogenous bacterial contamination. The ventral body walls were incised using a sterile scalpel to expose the body cavity. During the process of dissection, we found the two species of sea cucumbers have long guts with the whole length of 83.40±8.52 cm and 43.30±3.43 cm inH.leucospilotaandH.atra, respectively. In order to investigate the changes of the gut microbial community which were just ingested and the contents which were about to be discharged, the foregut (the most anterior 2-3-cm part of the gut) and hindgut contents (the most posterior 2-3-cm part of the gut)were separately collected in sterile freezing tubes(Gao et al., 2014a; Mf ilinge and Tsuchiya, 2016). The foregut samples ofH.leucospilotaandH.atrawere labeled as YQ1-YQ5 and HQ1-HQ5, respectively,and the hindgut samples were marked as YH1-YH5 and the HH1-HH5, respectively. The sediments were marked by DN1-DN5. All samples were stored at-80 °C for further analysis.
2.2 Method
2.2.1 DNA extraction and polymerase chain reaction(PCR) amplif ication
Omega Soil DNA Kit (Omega Bio-Tek, Norcross,GA, USA) was used to extract DNA from the gut content and sediment according to the manufacturer’s protocol.
PCR amplif ication of bacterial 16S rRNA gene hypervariable regions (V3-V4) was conducted using the universal primers, including 341F(5′-CCTAYGGGRBGCASCAG-3′) and 806R (5′-GGACTACNNGGGTATCTAAT-3′), with barcodes to allow sample multiplexing (Wang et al., 2018; Gao et al., 2019). A total of 30-μL PCR mixture contained 15 μL of Phusion®High-Fidelity PCR Master Mix(New England Biolabs), 0.2 μmol/L of forward and reverse primers, and 10-ng template DNA. Thermal cycling consisted of initial denaturation at 98 °C for 1 min, followed by 30 cycles of denaturation at 98 °C for 10 s, annealing at 50 °C for 30 s, elongation at 72 °C for 30 s and a f inal extension step at 72 °C for 5 min. PCR products were mixed in equidensity ratios and purif ied with GeneJETTM Gel Extraction Kit (Thermo Scientif ic).
2.2.2 Library preparation and sequencing
Sequencing libraries were generated using Ion Plus Fragment Library Kit 48 rxns (Thermo Scientif ic)following the manufacturer’s instructions. The library quality was assessed on the Qubit@ 2.0 Fluorometer(Thermo Scientif ic). The library was sequenced on an Ion S5TM XL platform, and 400-bp/600-bp singleend reads were generated.
2.2.3 Statistical and bioinformatics analysis
The data were analyzed using Statistical Package for the Social Sciences (SPSS) 19.0. All values were presented as the means±standard error (mean±SE).The level of statistical signif icance was determined using thet-test. The statistical signif icance was set atP<0.05.
Single-end reads were assigned to samples based on their unique barcode and truncated by cutting offthe barcode and primer sequences. Quality f iltering on the raw reads was performed under specif ic f iltering conditions according to the Cutadapt (V1.9.1, http://cutadapt.readthedocs.io/en/stable/) quality-controlled process (Martin, 2011). The reads were compared with the reference database (Silva database, https://www.arb-silva.de/) using UCHIME algorithm (http://www.drive5.com/usearch/manual/uchime_algo.html)to detect chimera sequences, and the chimera sequences were then removed (Edgar et al., 2011).
Sequences analysis was performed by Uparse software (Uparse v7.0.1001, http://drive5.com/uparse/; Edgar, 2013). The sequences with ≥97%similarity were assigned to the same operational taxonomic units (OTUs). The representative sequence for each OTU was screened for further annotation.For each representative sequence, the Silva Database(https://www.arb-silva.de/) was used based on the Mothur algorithm to annotate taxonomic information.
Alpha diversity is applied in analyzing complexity of species diversity for a sample through 4 indices,including the abundance-based coverage estimator(ACE), bias-corrected Chao1 richness estimator, and the Shannon and Simpson diversity indices. were calculated. All the analyses were performed using the Mothur program (http://www.mothur.org). The microbial community structures in diff erent samples were calculated by Quantitative Insights Into Microbial Ecology (QIIME2) software (version 1.7.0). Non-metric multidimensional scaling (NMDS)analysis was performed with the R software (Version 2.15.3). Unweighted Pair-group Method with Arithmetic Means (UPGMA) Clustering was performed using average linkage and conducted by QIIME2 software (Version 1.7.0).
3 RESULT
A total of 2 002 434 optimized reads with an average length of 408 bp were obtained from all the samples. The optimized read numbers for each sample ranged from 80 023 to 80 174, with a mean average of 80 097±9. The Good’s coverage of all samples was between 98.9% and 99.5%, which showed that the sequencing depth was adequate to cover most microorganisms.
3.1 Richness and diversity
A total of 5 584 OTUs were obtained from 25 samples based on a 97% threshold. The foregut content samples from each of the f iveH.leucospilota(YQ1-YQ5) contained 1 145-1 592 OTUs, and the hindgut contents (YH1-YH5) from each of the f ive sea cucumbers contained 940 to 1 427 OTUs. The foregut (HQ1-HQ5) and hindgut contents (HH1-HH5) from each of the f iveH.atracontained 1 016-2 134 OTUs, and 1 101-1 411 OTUs, respectively.The f ive surface sediments (DN1-DN5) contained 2 497-2 635 OTUs.
Of these OTUs, 369, 147, 109, 177, and 1 088 OUT were detected in YQ, YH, HQ, HH, and DN,respectively (Fig.1). A total of 995 OTUs were shared by all the gut content samples and their surrounding sediments (Fig.1).
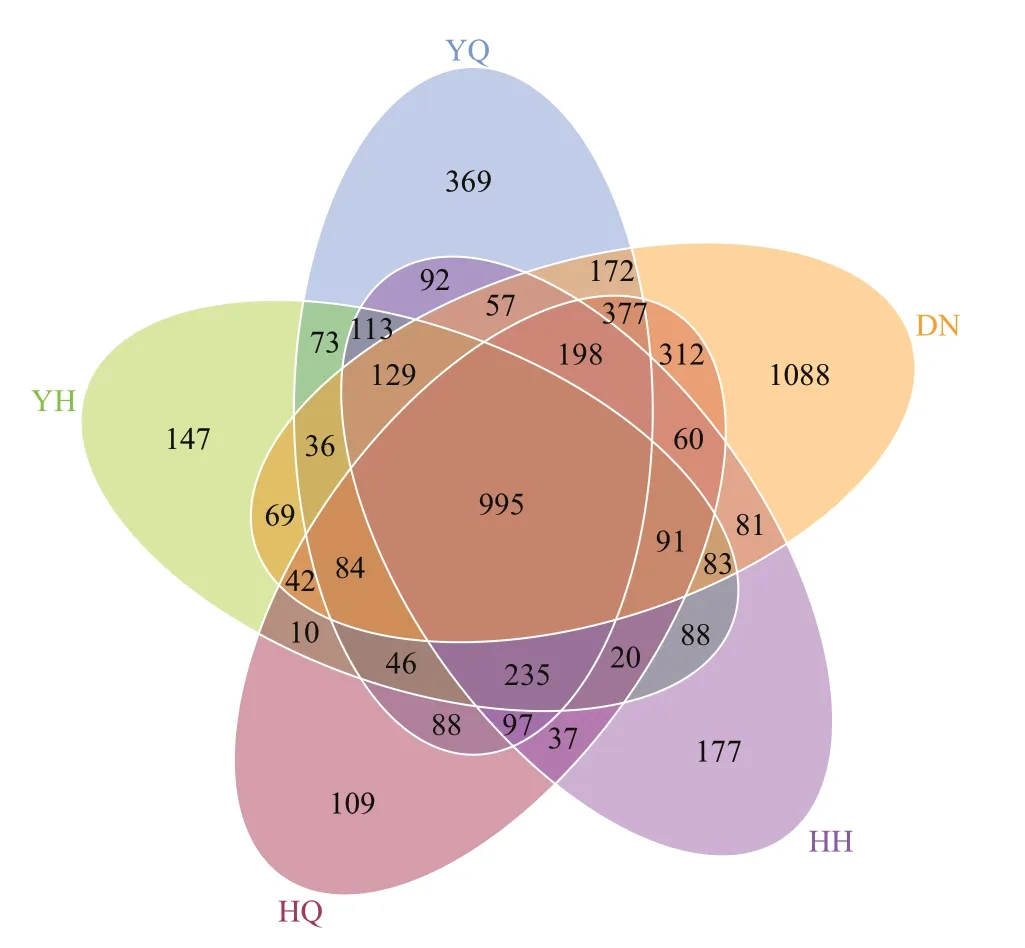
Fig.1 Venn diagram of core OTUs among the foregut(YQ), hindgut (YH) of H. leucospilota, the foregut(HQ), hindgut (HH) of H. atra and the surrounding sediments (DN)
The bacterial community richness was calculated by estimating the number of OTUs based on the ACE and Chao1 values at the 3% dissimilarity level. The ACE and Chao1 values in the sediment samples were signif icantly higher than the values in all the foregut and hindgut contents of two species of sea cucumbers(Fig.2a & b;P<0.05), indicating that the bacterial richness in the sediment samples was higher than that in the gut content samples. However, there were no signif icant diff erences while comparing between the same parts of the guts of the two sea cucumber species(P>0.05).
The Shannon and Simpson diversity indices in the sediment samples were higher than the indices in all the gut content samples (Fig.2c & d;P<0.05), but there were no signif icant diff erences while comparing between the same parts of gut of the two sea cucumber species (P>0.05). The results suggested that the bacterial diversity in the habitat sediments was higher than that in the gut contents of the sea cucumbers, but not clear diff erence between the gut contents of the two sea cucumbers (P>0.05).
3.2 Bacterial community structure in gut contents and sediments
Most of the reads (97.6%-99.8%) could be classif ied at the phylum level in all samples. Of these,a total of 39 and 31 diff erent phyla were identif ied in the foregut and hindgut contents ofH.leucospilota. In the foregut and hindgut contents ofH.atra, the bacteria could be separately classif ied into 36 and 35 diff erent phyla. The identif ied bacteria community in the ambient sediments was 40 phyla.
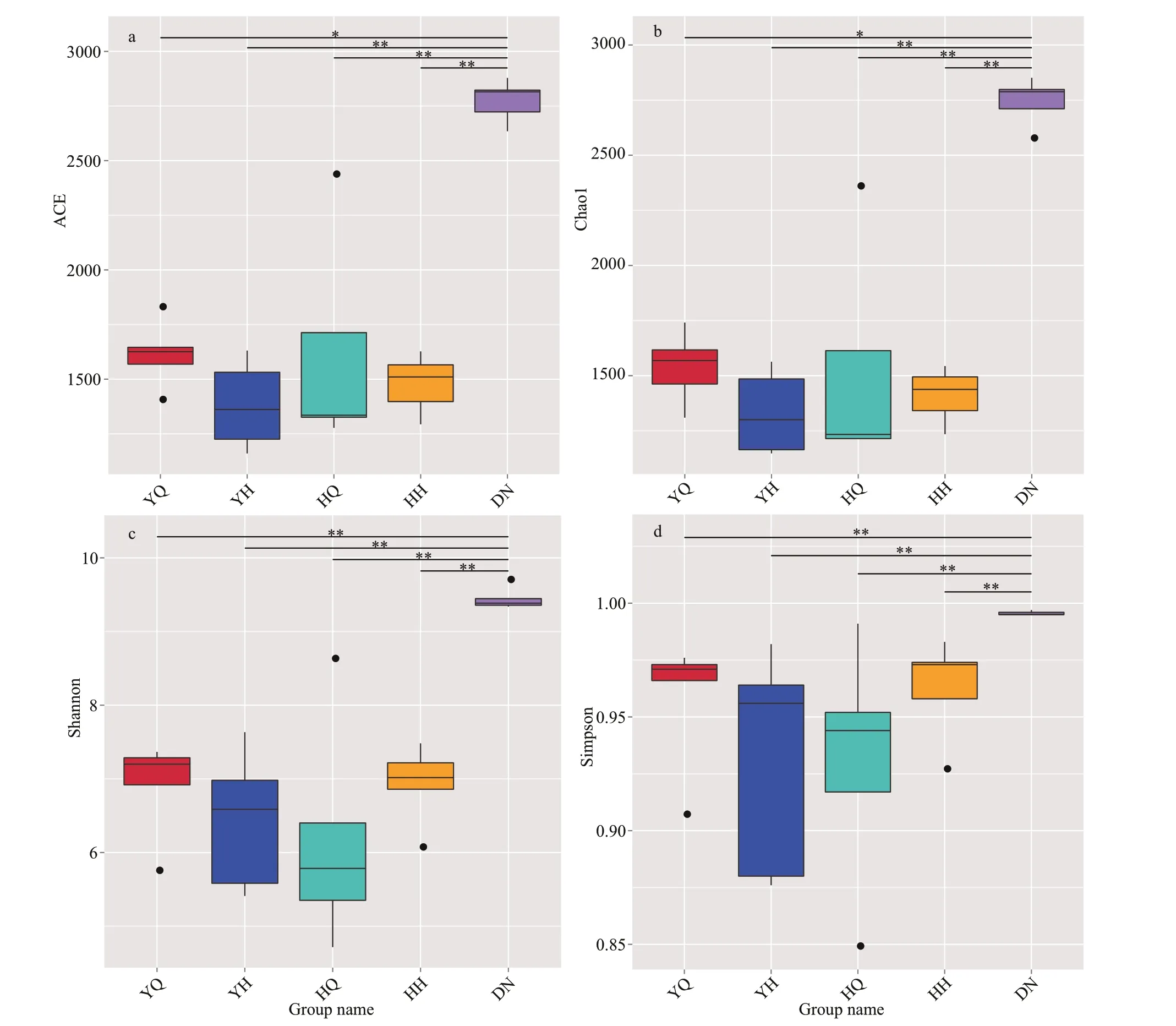
Fig.2 Alpha-diversity of microbial communities in the foregut (YQ), hindgut (YH) of H. leucospilota, the foregut (HQ),hindgut (HH) of H. atra and the surrounding sediments (DN): ACE (a), Chao1 (b), Shannon index (c), and Simpson index (d)
The ten most abundant phyla of the diff erent samples are listed in Fig.3. Proteobacteria was the predominant phylum, showing 45.69%±8.61%,70.09%±4.03%, 45.88%±5.38%, and 55.19%±0.79%reads in the YQ, YH, HH, and DN libraries,respectively. In the HQ samples, the relative content of Proteobacteria was 20.33%±5.75%, while Bacteroidetes was the predominant phylum with a relative content of 34.98%±5.52%. There was no signif icant diff erence in the abundance of Proteobacteria among the foregut and hindgut contents inH.leucospilotaand sediment samples (P>0.05).The abundance of Proteobacteria in the hindgut content ofH.atrawas signif icantly lower than that in surrounding sediments and foregut content (P<0.05).
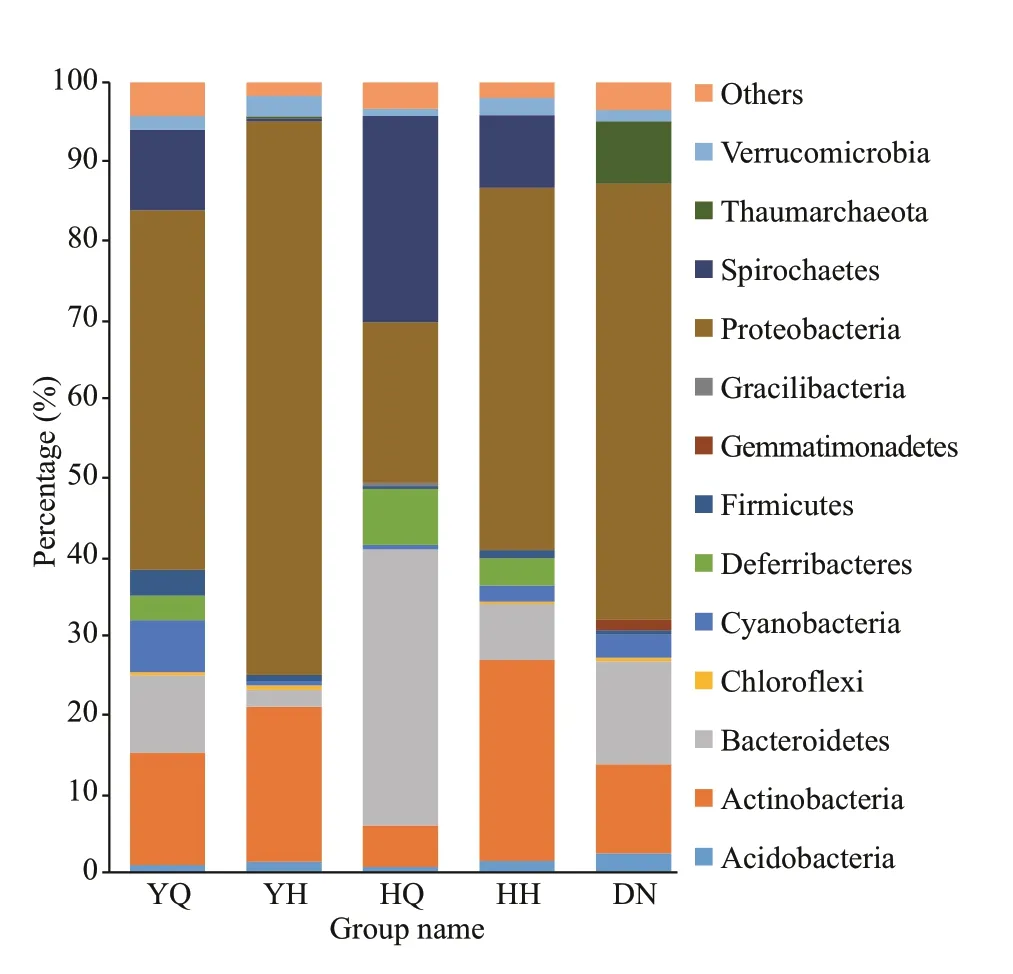
Fig.3 The relative abundance of the 10 most abundant phyla
While comparing the bacterial abundance in foregut contents with that in sediments at the phylum level, the contents of Thaumarchaeota, Acidobacteria,Gemmatimonadetes, Latescibacteria, Fusobacteria,Nitrospirae, and Calditrichaeota in the foregut ofH.leucospilotawere signif icantly lower than those in sediments (P<0.05; Fig.4a). The contents of Proteobacteria, Thaumarchaeota, Acidobacteria,Gemmatimonadetes, Latescibacteria, Fusobacteria,Nitrospirae, and Calditrichaeota in the foregut ofH.atrawere signif icantly lower than those in sediments (P<0.05), but the contents of Bacteroidetes and Spirochaetes in the foregut ofH.atrawere signif icantly higher than those in sediments (P<0.05;Fig.4b). The relative abundance of Bacteroidetes,Thaumarchaeota, Acidobacteria, Gemmatimonadetes,Latescibacteria, Fusobacteria, Nitrospirae, and Calditrichaeota in the hindgut ofH.leucospilotawere signif icantly lower than those in sediments, but the contents of Proteobacteria and Verrucomicrobia in the hindgut ofH.leucospilotawere signif icantly higher than those in sediments (P<0.05; Fig.4c). The contents of Thaumarchaeota, Acidobacteria,Gemmatimonadetes, Chlorof lexi, Planctomycetes,Latescibacteria, Fusobacteria, Nitrospirae, and Calditrichaeota in the hindgut ofH.atrawere signif icantly lower than those in sediments (P<0.05),but the contents of Actinobacteria and Dadabacteria in the hindgut ofH.atrawere signif icantly higher than those in sediments (P<0.05; Fig.4d).
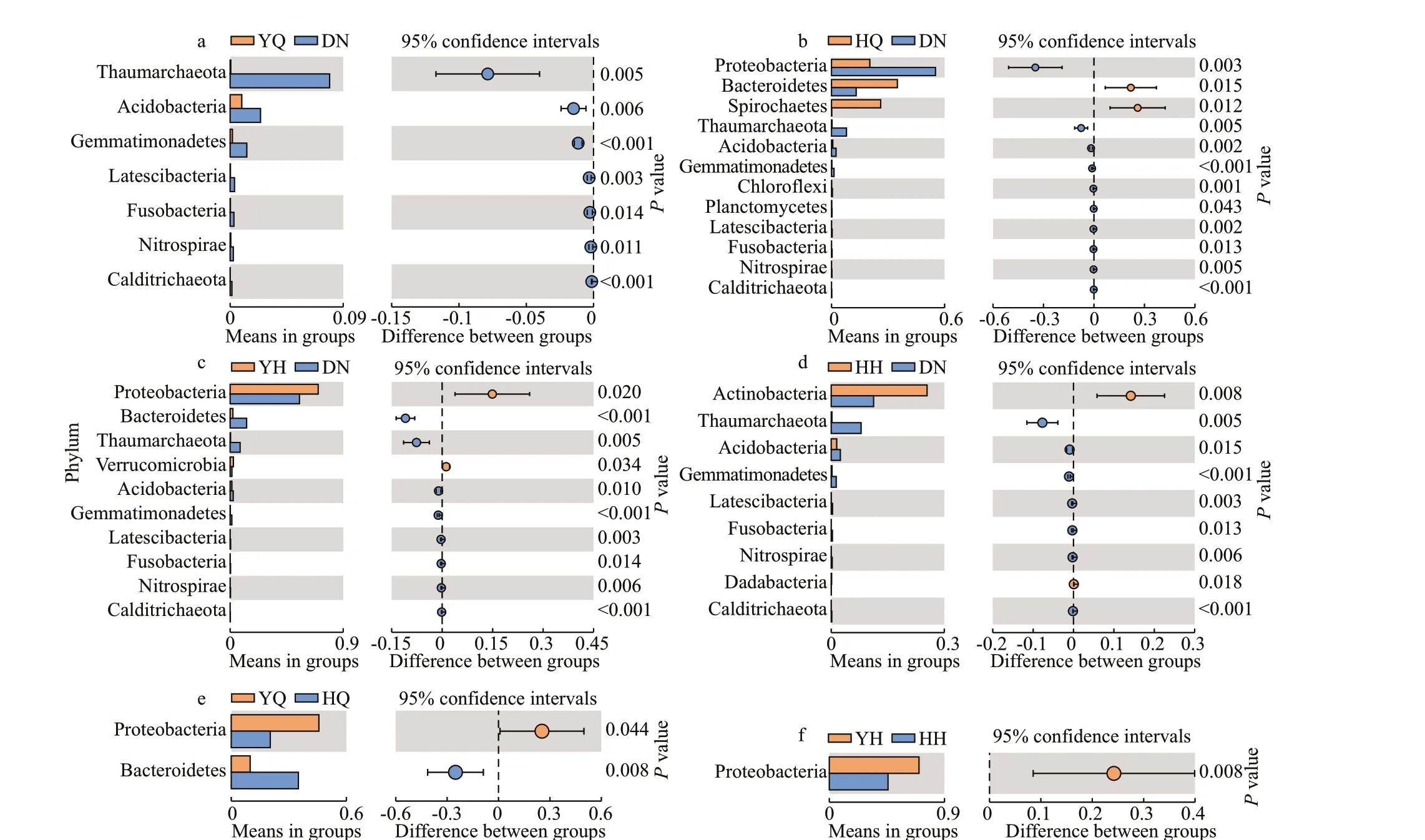
Fig.4 Identif ied diff erentially abundant phyla between YQ and DN (a), HQ and DN (b), YH and DN (c), HH and DN (d), YQ and HQ (e), YH and HH (f) ( P <0.05)
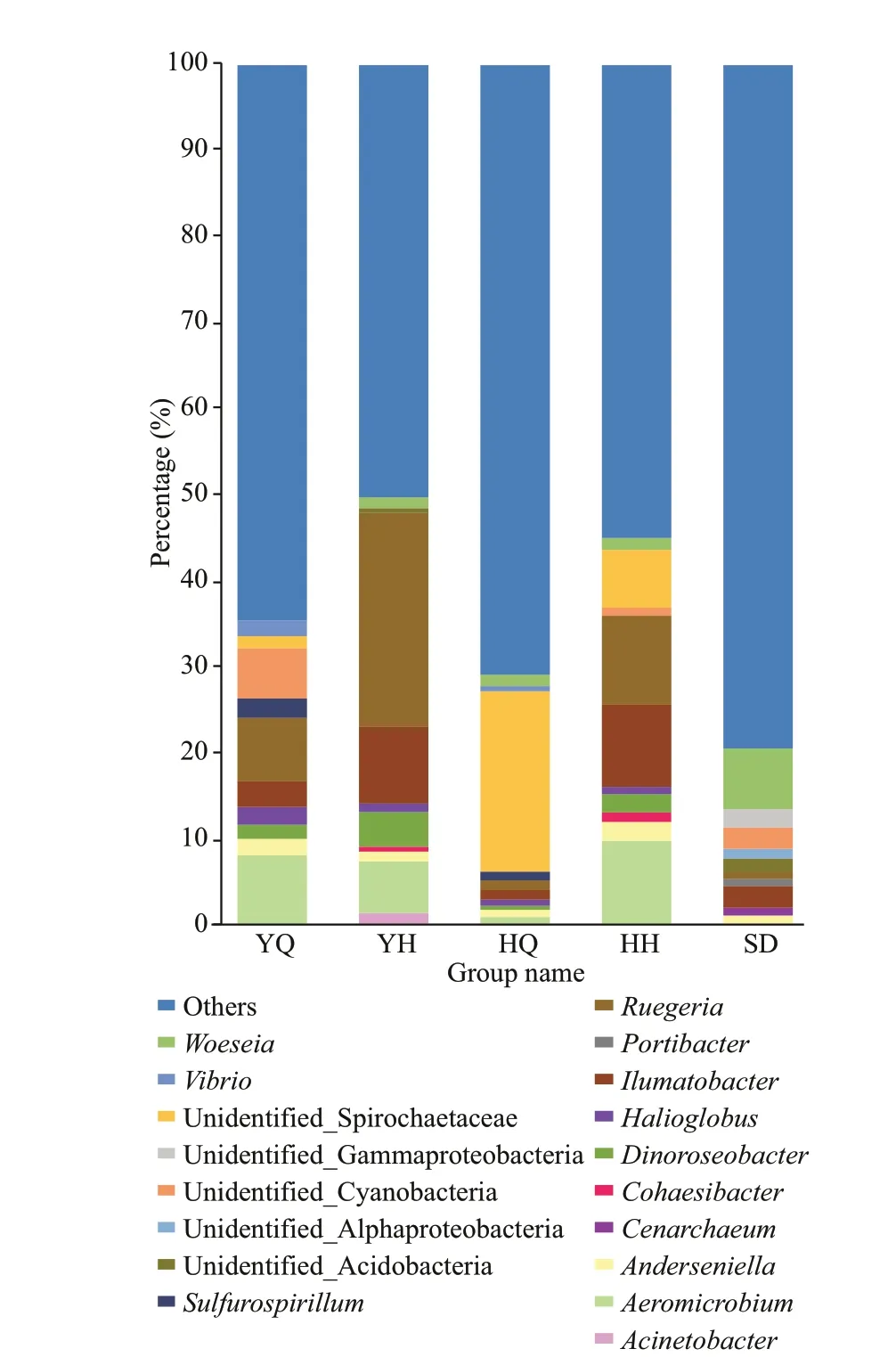
Fig.5 The relative abundance of the 10 most abundant genera
Most of the taxa had no signif icant diff erences while comparing the bacterial abundance in the same part between the two sea cucumber species. The content of Proteobacteria in the foregut ofH.leucospilotawas signif icantly higher than that in the foregut ofH.atra(P<0.05), and Bacteroidetes content in the foregut ofH.leucospilotawas signif icantly lower than that in foregut ofH.atra(P<0.05; Fig.4e). As for the hindgut, only the content of Proteobacteria inH.leucospilotawas signif icantly higher than that inH.atra(P<0.05; Fig.4f).
At the genus level, a large proportion (21.5%-83.2%) of the reads could not be classif ied. The ten most abundant genera in the diff erent samples accounted for 35.39%, 49.73%, 29.09%, 45.01%, and 20.51% reads in the YQ, YQ, HQ, HH, and SD libraries, respectively (Fig.5). The most abundant genera in the foregut content libraries ofH.leucospilotawere sequences related toAeromicrobium(8.06%±7.34%),Ruegeria(7.40%±2.85%),unidentif ied Cyanobacteria (5.83%±3.27%),Ilumatobacter(2.97%±0.82%),Sulfurospirillum(2.25%±1.37%),Halioglobus(2.10%±1.03%),Anderseniella(1.91%±1.08%),Vibrio(1.84%±1.34%),Dinoroseobacter(1.63%±0.67%), and unidentif ied Spirochaetaceae (1.40%±0.88%). In the hindgut libraries ofH.leucospilota, the most abundant genera(relative abundance >1%) wereRuegeria(24.87%±6.25%),Ilumatobacter(8.91%±3.35%),Aeromicrobium(6.00%±2.47%),Dinoroseobacter(4.03%±1.22%),Acinetobacter(1.36%±1.21%),Woeseia(1.27%±0.37%),Anderseniella(1.13%±0.28%), andHalioglobus(1.01%±0.18%).The most abundant genera in the foregut content libraries ofH.atrawere sequences related to unidentif ied Spirochaetaceae (20.99%±5.80%),Woeseia(4.03%±1.22%),Ruegeria(1.35%±0.67%),Ilumatobacter(4.03±1.22%), andSulfurospirillum(1.01±0.42%). In the hindgut libraries ofH.atra, the most abundant genera (relative abundance >1%) wereRuegeria(10.36%±5.05%),Aeromicrobium(9.75%±4.46%),Ilumatobacter(9.57%±1.27%),unidentif ied Spirochaetaceae (6.77%±4.23%),Anderseniella(2.20%±0.32%),Dinoroseobacter(2.14%±0.49%),Woeseia(1.38%±0.41%), andCohaesibacter(1.09±0.19%). In the sediment libraries,Woeseia(7.06%±0.97%),Ilumatobacter(2.53%±0.32%), unidentif ied Cyanobacteria(2.50%±0.79%), unidentif ied Gammaproteobacteria(2.14%±0.15%), unidentif ied Acidobacteria(1.62%±0.18%), unidentif ied Alphaproteobacteria(1.09%±0.44%) andAnderseniella(1.01%±0.10%)were the dominant genera (relative abundance >1%).
Among the dominant genera, reads related to the generaAnderseniella,Ilumatobacter, andRuegeriawere detected in all the gut contents and sediment libraries. In the hindgut of the two species of sea cucumber, the abundance of genusRuegeriawas very high, 24.87%±6.25% and 10.36%±5.05% inH.leucospilotaandH.atra, respectively, and theRuegeriaabundance in the hindgut ofH.leucospilotawas signif icantly higher than that in the other samples (P<0.05).
3.3 Relationships among the bacterial communities from the gut content and the sediment samples
NMDS analysis and UPGMA tree were performed to assess the relatedness of the microbial community composition among diff erent samples (Figs.6 & 7).The analyses indicated that gut microbiota in the foregut and hindgut ofH.atraand sediment were clustered separately into three groups, which showed that, forH.atra, the foregut, hindgut, and sediment had obviously diff erent characteristics of bacterial communities. The NMDS analysis showed that the bacterial community composition of the foregut inH.leucospilotawas overlapped with the characteristics of the hindgut samples of two sea cucumber species and the foregut samples ofH.atra. The bacterial community of sediment was diff erent from all the foregut and hindgut bacterial communities in bothH.atraandH.leucospilota. The hindguts ofH.atraandH.leucospilotahad comparatively similar bacterial communities.

Fig.6 Nonmetric multidimensional scaling (NMDS)plot based on Bray-Curtis distance showing the relatedness of the bacterial community composition between the diff erent samples
4 DISCUSSION
As typical deposit-feeding or detritus-feeding marine invertebrates, the feeding and nutrition of aspidochirotid sea cucumbers have received considerable attention. In this study, we investigated the bacterial community composition in diff erent parts of gut of two commercially exploited tropical sea cucumbers speciesH.atraandH.leucospilotaand in their surrounding sediments, in order to assess the sea cucumber’s feeding strategy and the eff ects on the sedimentary microbiota.
4.1 Dominant bacteria in the gut contents
A total of 46 diff erent phyla were identif ied from the gut contents of two species of sea cucumbers, and their ambient sediments showed 40 phyla. Of these,Proteobacteria was the predominant phylum in all the gut content samples ofH.leucospilota, hindgut contents ofH.atraand sediments. In previous studies,Proteobacteria was found to be the predominant phylum in the gut contents ofApostichopusjaponicus(Gao et al., 2014a; Wang et al., 2018), marine water,and sediments (Paliaga et al., 2017; Wang et al., 2019;Custodio et al., 2020).
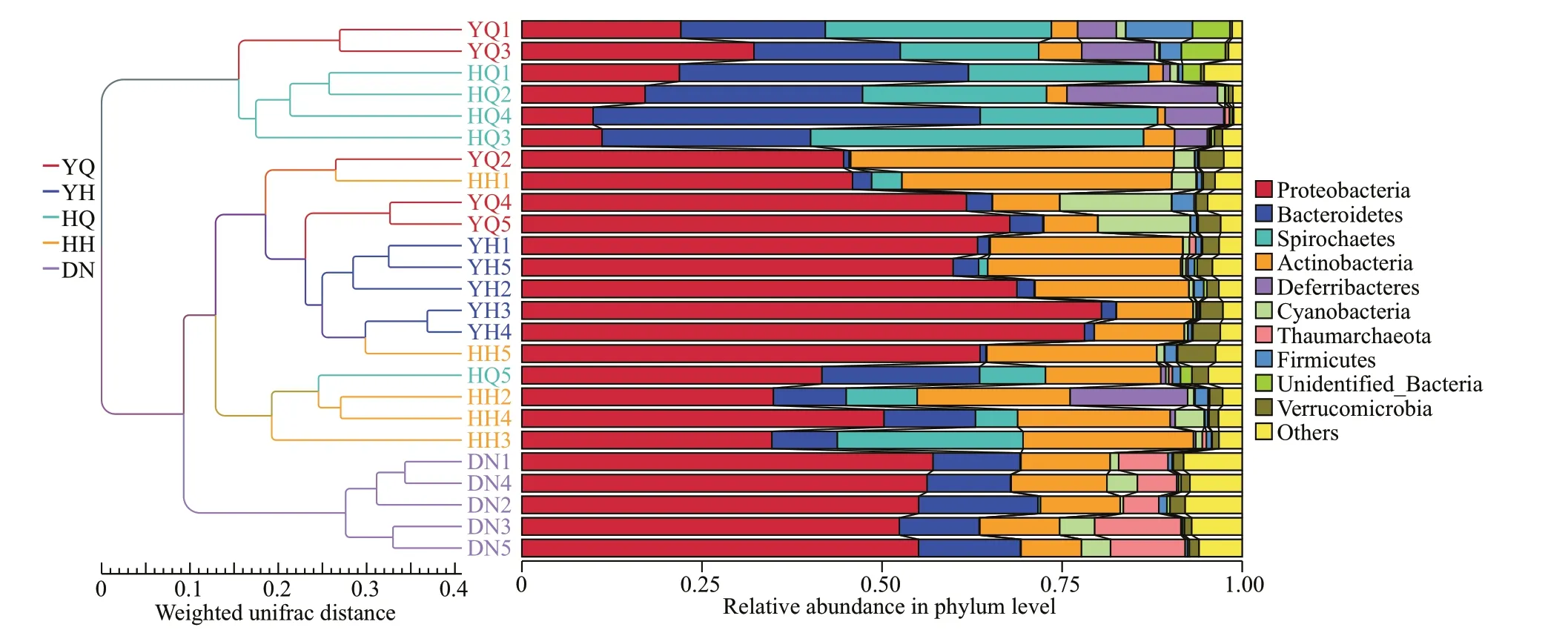
Fig.7 UPGMA tree showing the similarity of bacterial community structures among the diff erent samples
Ruegeriawas the predominant bacteria in the hindgut content of two species of sea cucumbers in our study, and the content in the hindgut ofH.leucospilotawas higher than that in the other samples (P<0.05). The genusRuegeriawas f irst reported by Uchino et al. (1998) based on the reclassif ication ofAgrobacteriumgelatinovorumandRoseobacteralgicolaasRuegeriagelatinovoraandR.algicola, respectively. Till now, the genusRuegeriacomprises about 18 species which have been named,and most of these are isolated from marine environments, including marine sediment, seawater,seashore, sea squirt, and oyster (Arahal et al., 2018;Kim et al., 2019; Baek et al., 2020). The common characteristics of the genusRuegeriaare nonsporulating, non-motile, ovoid, or rod-shaped cells that contain Q-10 as a predominant respiratory quinone (Baek et al., 2020). Species of the genus are chemoorganotrophic aerobes, slightly halophilic and mesophilic, and none of them can synthesize bacteriochlorophylla(Arahal et al., 2018). The content ofWoeseiain sediments was signif icantly higher than that of two species of sea cucumbers(P<0.001). TheWoeseiabacteria are Gram-negative,rods or bent rods, non-motile, and facultatively anaerobic, and require salt to grow.Woeseiashowed an unusually high abundance in marine sediments,including coastal sediments, due to the broad range of energy-yielding metabolisms (Muβmann et al., 2017).
Vibriohas been found widely distributed in the digestive tract of sea cucumbers, such asA.japonicus(Sun and Chen, 1989; Gao et al., 2014a),H.atra(Ward-Rainey et al., 1996), andH.leucospilota(Zhang et al., 2012). In the present study,Vibriowas found in all the gut contents, sediment samples, and foreguts of two sea cucumbers species. AlthoughVibrioisolates are commonly believed to be pathogenic for aquatic animals, a lot of avirulent strains have been used as probiotics (Rahiman et al.,2010; Silva-Aciares et al., 2011; Pandiyan et al.,2013).Vibriosp. V33 isolated from healthy sepia showed intense antagonistic activity against pathogenicVibriosplendidusVs (Liu et al., 2017). A total of 13 species ofVibrioisolated from the digestive tract ofH.leucospilotashowed amylase and alginatelyase activities (Zhang et al., 2012). Chi et al. (2014)isolatedV.tasmaniensisfrom the gut ofA.japonicusand considered this strain as potential probiotics inA.japonicusafter demonstrating its extra-cellular amylase, lipase, and protease production capacities and a series of dietary supplementation tests.
4.2 Comparison of gut microbe composition in H. leucospilota and H. atra
While comparing the bacterial abundance in the same gut part between the two species of sea cucumbers, most of the taxa had no signif icant variations. In the foregut contents, only Proteobacteria and Bacteroidetes had signif icantly diff erent ranges betweenH.leucospilotaandH.atra. NMDS analysis showed the foregut content samples ofH.atragrouped intensively, yet the bacterial community composition of the foregut inH.leucospilotawas overlapped with the characteristics of the hindgut samples of two sea cucumber species and the foregut samples ofH.atra, which indicated microbial food sources of sea cucumberH.leucospilotahas much more diff erences among individuals thanH.atra. In other words,H.atramight have a stronger feeding preference thanH.leucospilota.
In the hindgut contents,H.atraandH.leucospilotahad comparatively similar bacterial communities,with only the content of Proteobacteria inH.leucospilotadiff ering signif icantly fromH.atra.Similar gut sediment fatty acid composition and concentration, including fatty acid biomarkers for bacteria, were found inH.atraandH.leucospilota,and the authors considered that ingestion of the same type of organic material was the primary reason(Mf ilinge and Tsuchiya, 2016). In this study, we speculated that the high similarity of bacterial community composition in hindgut contents between two sea cucumbers species was probably due to escaping digestion of the same type of microbes.
4.3 Relationship between bacterial communities in the foregut and the sediments
It has long been focused on whether deposit-feeding sea cucumbers could feed selectively. Some reports have claimed that sea cucumbers feed selectively,particularly with respect to particle size (Hauksson,1979; Roberts, 1979; Zhao, 2010; Xue et al., 2019),bacterial biomass, and community composition(Moriarty, 1982; Sun and Chen, 1989; Gao et al.,2014a, b), and organic matter content of sediments(Moriarty, 1982; Paltzat et al., 2008; Zhao, 2010; Xue et al., 2019). In most studies, it is usual to compare specif ic parameters of the foregut content with the corresponding parameters in the ambient sediment to describe the selectivity in sediment feeders.
Beside of selective feeding, microf lora attaching to the gut wall or microbial reproduction in the gut might also be the possible sources of the distinct bacterial community composition in the adjacent sediments and foregut contents of sea cucumbers. In this study, we tried to minimize the two impact factors while extracting the samples. Firstly, merely the contents in the most anterior part of the foregut (about 2-3 cm,though the whole gut length inH.leucospilotaandH.atrawas 43.30±3.43 cm and 83.40±8.52 cm,respectively) were extracted as the foregut contents in order to reduce the eff ect of specif ic microbial reproduction in guts. We considered that there would not be much time for bacteria to grow there (Moriarty,1982; Gao et al., 2014a). Secondly, the gut contents were squeezed out carefully, making certain that contents adhering to gut issue were excluded, so as to lessen the possible autochthonous bacteria sources. In this study, the bacterial community composition between the foregut content of two species of sea cucumbers and the surrounding sediments was compared, which indicated diff erent bacterial composition between the sediment and foregut samples inH.leucospilotaandH.atra. Therefore, we speculate that the diff erent bacterial communities in the foregut content and sediments may mainly result from selective feeding. Moriarty (1982) found that the bacterial biomass and organic carbon levels in the foregut ofH.atrawere two to three-fold greater than in the surrounding sediment, and indicated that the animals selected sediment fractions that contained more bacteria and organic matters. We also investigated the sediment characteristics ingested byH.atrain a typical tropical coral reef in the South China Sea, and the results showed that the organic matter content and grain size ratio in the digestive tract ofH.atrawere signif icantly diff erent from those in environmental sediments (Xue et al., 2019). Fatty acids, including saturated, monounsaturated, polyunsaturated, and branched fatty acids, were signif icantly higher in the foregut ofH.atraandH.leucospilotathan those in ambient sediments, suggesting that both species selectively ingested nutrient-rich particles (Mf ilinge and Tsuchiya, 2016). As it is impossible to identify the microbe community diversity or abundance in diet for any species of animals, we supposed that the clearly diff erent bacterial composition between foregut contents and the surrounding sediments might be due to their selective feeding of sediment patches.
Highly selective feeding has been observed in various species of sea cucumbers from temperate to tropical habitats, regardless of shallow-water ones or deep-sea ones (Khripounoff and Sibuet, 1980; Sloan and von Bodungen, 1980; Hammond, 1983; Billett et al., 1988; Amaro et al., 2009; Gao et al., 2014b), and the objects selected including grain size of particles,organic content, sediment patches, and so on. Another question is how the deposit-feeding sea cucumbers selectively feed. Holothuroids take in food by means of their oral tentacles. Histological examination indicated that the tentacle structure, including the size of nodules or nodule groups, inter-papillar spaces,and mucous secretion ability of the nodules were responsible for the physical selection for certain particle sizes (Fankboner, 1978; Roberts, 1979; Foster and Hodgson, 1996). As for the mechanisms of selective feeding of other parameters, such as organic contents and composition in sediments, it is theoretically required well developed sensory organs for sediment quality judging. Foster and Hodgson(1996) found that, in several species of sea cucumbers,the nodules at the terminal of each tentacle have lengthened epithelial cells with papillae comprising cilia, which are distinct from other tentacle parts. The papillae have been regarded as sensory receptors for feeding (Fankboner, 1978; Smith, 1983).
4.4 Impact on sediment bacterial community by feeding of sea cucumbers
Bacteria play an important role in coral reef processes consisting of organic matter decomposition,nutrient recycling, and so on. Sea cucumbers are prominent deposit-feeders as they can rework considerable quantity of sediments (Bonham and Held, 1963; Mangion et al., 2004), and it has long been proved that bacteria were one kind of important food sources for sea cucumbers (Moriarty, 1982;Uthicke, 1999; Gao et al., 2010; MacTavish et al.,2012). Therefore, sea cucumber’s feeding activity may inf luence the bacterial community diversity and biomass in sediments. As it is diffi cult to f ind and collect the fresh feces in situ, we collected the posterior 2-3 cm of the gut contents as the hindgut samples, and we assumed that this section of gut content had the most similarity with the feces which would be passed out. In the present study, NMDS analysis and UPGMA tree both showed the bacterial community structure in hindgut content of the two species of sea cucumbers diff ered clearly from sediments. In details, the relative contents of ten phyla of bacteria including Proteobacteria, Bacteroidetes,Thaumarchaeota, Acidobacteria, Gemmatimonadetes,Latescibacteria, Fusobacteria, Nitrospirae,Calditrichaeota, and Verrucomicrobia in the hindgut ofH.leucospilotawere signif icantly diff erent from those in sediments, and the relative contents of phyla Actinobacteria, Thaumarchaeota, Acidobacteria,Gemmatimonadetes, Chlorof lexi, Planctomycetes,Latescibacteria, Fusobacteria, Nitrospirae,Dadabacteria, and Calditrichaeota in the hindgut ofH.atrawere signif icantly diff erent from those in sediments. The results indicated that marked changes in sediment bacterial composition occurred during gut passage of the sediment particles in both sea cucumber species. Clearly diff erent bacterial biomass between the hindgut contents ofH.atraand sediments were also observed in Heron Island, Great Barrier Reef (Moriarty, 1982). When sediment bacterial biomass is low, feeding activity ofH.tubulosaresults in a net increase in bacteria during the gut passage,while comparatively high bacterial biomass in sediments leads to a net bacterial removal (Amon and Herndl, 1991). In conclusion, to some extent, depositfeeding sea cucumbers, includingH.leucospilotaandH.atra, could actually inf luence sediment bacterial community structure through their feeding activities.
5 DATA AVAILABILITY STATEMENT
The datasets generated during and/or analyzed during this study are available from the corresponding author on reasonable request.
 Journal of Oceanology and Limnology2022年1期
Journal of Oceanology and Limnology2022年1期
- Journal of Oceanology and Limnology的其它文章
- Spatial diff erence in net growth rate of Yesso scallop Patinopecten yessoensis revealed by an aquaculture ecosystem model*
- Comparative transcriptomic analysis of Macrobrachium nippon ense in response to Aerom onas veronii or Staphylococcus au reus infection*
- Dietary intake of bamboo vinegar and charcoal powder (BVC)enhances resistance of African catf ish Clarias gariepinus to bacterial pathogen*
- Cloning of catalase gene and antioxidant genes in Scophthalmus maximus response to metalloprotease of Vibrio anguillarum stress*
- Taxonomy and regeneration of a newly recorded Polychaete Capitella teleta (Annelida, Capitellidae) in the coastal water of Shandong, China*
- Five new records of Xanthidae (Crustacea: Brachyura) from Hainan Island, China*