
Comparative transcriptomic analysis of Macrobrachium nippon ense in response to Aerom onas veronii or Staphylococcus au reus infection*
2022-03-05 09:36:40ChongHANZhipengZHANGQiangLIQinghuaCHENJianrongHUANG
Chong HAN , Zhipeng ZHANG , Qiang LI , Qinghua CHEN , Jianrong HUANG ,2,**
1 State Key Laboratory of Biocontrol, Institute of Aquatic Economic Animals and Guangdong Provincial Key Laboratory for Aquatic Economic Animals, School of life Sciences, Sun Yat-Sen University, Guangzhou 510275, China
2 Southern Marine Science and Engineering Guangdong Laboratory (Zhuhai), Zhuhai 519082, China
3 School of life Sciences, Guangzhou University, Guangzhou 510006, China
4 South China Institute of Environmental Science, MEE, Guangzhou 510610, China
Abstract M acrobrachium nipponense is an economically important freshwater prawn that is often threatened by many aquatic pathogens. In this study, comparative transcriptomic analysis was f irstly used to explore the transcriptional response of M. nipponense to Aeromonas veronii or Staphylococcus aureus stimulation. A total of 400.19 million clean reads were obtained and assembled into 56 944 unigenes with an average length of 1 253 bp. A total of 1 857 diff erentially expressed genes were found after A. veronii infection, including 677 genes that were up-regulated and 1 180 genes that were down-regulated, while 1 061 signif icant diff erentially expressed genes were identif ied after S. aureus infection, including 390 upregulated and 671 down-regulated genes. Many immune-related genes including Spaetzle, prophenoloxidase activating factor, C-type lectin, anti-lipopolysaccharide factor, and inhibitor of apoptosis 2 protein were commonly up-regulated after A. veronii or S. aureus infection. This study will enrich our understanding of the immune response to gram-positive and gram-negative bacteria infection in crustaceans.
Keyword: Macrobrachium nipponense; diff erentially expressed genes; Aeromonas veronii; Staphylococcus aureus; immune response
1 INTRODUCTION
The oriental river prawnMacrobrachium nipponense, belonging to thePalaemonidaefamily of decapod crustaceans, is an important aquaculture prawn and is widely reared in China, Japan, and other South-East Asian countries (Cai and Dai, 1999; Cai and Ng, 2002). Owing to the short breeding period and high economic value, this species is popular in aquaculture and has become one of the four major freshwater shellf ish species cultivated in China (Yao et al., 2004). However, various diseases caused by bacteria inM.nipponensehave placed signif icant constraints on aquaculture production and have led to severe economic losses in prawn farming (Yao et al.,2004). Thus, it is urgent to understand the immune defence mechanism ofM.nipponenseat the molecular level after bacterial infection.
Aeromonas veronii, a motile, mesophilic aeromonad, is a gram-negative pathogen that consists of motile strains and grows well at optimal growth temperatures between 35 and 37 °C (Janda and Abbott, 2010). This pathogen has been characterized as a “highly virulent pathogen” that can cause motile aeromonad septicaemia in a wide range of species, from invertebrates to mammals(Havixbeck et al., 2017).A.veroniihas had devastating eff ects on the aquaculture industry and has led to disastrous economic losses for farmers(Vazquez-Juarez et al., 2005; Sahoo et al., 2008;Zhang et al., 2017).A.veroniihas been reported to cause motile aeromonad septicaemia in many cultured species, includingLitopenaeus vannamei(Zhang et al., 2017),Eriocheir sinensis(Fang et al.,2008), andProcambarus clarkii(Jiang et al., 2016).Fang et al. (2008) isolated one dominant bacterial strain, AVZ01, from sickL.vannameiand identif iedA.veroniias the pathogen via in vitro tests, indicating thatA.veroniimight also lead to crustacean death in the aquaculture industry.
Staphylococcus aureusis a gram-positive cocci bacterium that can invade host cells and persist intracellularly for various durations in cell culture models (Garzoni and Kelley, 2009). Pathogenic strains often promote infections by producing a large repertoire of virulence factors such as secreted toxins,potent haemolysins and leukotoxins (Otto, 2014).Toxins damage biological membranes and inhibit the complement cascade or prevent recognition by host defences. Among haemolysins and leukotoxins, some represent powerful weapons against bacterial elimination through the innate host defence system(Wang et al., 2008).S.aureusis a global pathogen of species ranging from mammals to invertebrates and can induce an innate immune response in invertebrates such asHaliotis diversicolor(Wang et al., 2008) andM.nipponense(Pan et al., 2019).
Previous studies have given a few introductions on the anti-bacterial immunity ofM.nipponense. After challenge withAeromonas hydrophila, the expression of anti-lipopolysaccharide factors (ALFs) and extracellular copper/zinc superoxide dismutase(ECSOD) in the hepatopancreas were increased responsively (Xiu et al., 2013; Wang et al., 2015).Upon infected byVibrio parahaemolyticusandS.aureus, the expression levels of Toll1 in gills were signif icantly up-regulated (Pan et al., 2019). After infection withA.veronii, M-type lectin was increased signif icantly while L-type lectin was signif icantly down-regulated in intestine (Xiu et al., 2015a).Besides, a novel C-type lectin and two 2-transmembrane C-type lectin also participated in antibacterial activity and were up-regulated signif icantly afterS.aureus,A.veronii, orV.parahaemolyticusinfection (Xiu et al., 2016;Huang et al., 2019).
Recently, next-generation RNA-sequencing (RNASeq) techniques have been widely applied for both mapping and quantifying transcriptomes from plants and animals (Garber et al., 2011). RNA-Seq has been verif ied as an adequate tool for investigating the transcriptomic response of aquatic animals to ambient stress (Xia et al., 2013). Immune-related transcriptomes have been characterized in many aquatic animals such asSinonovacula constricta(Zhao et al., 2017),Mytilus chilensis(Núñez-Acuña and Gallardo-Escárate, 2013) andM.nipponense(Xu et al., 2016). RNA-Seq ofRuditapes philippinaruminfected byVibrio anguillarumidentif ied mRNAs as important eff ectors in the intricate host-pathogen interaction network (Ren et al., 2017). It was reported that the ability to respond to pathogen stimulation of an organism depends on its ability to initiate changes in gene expression (Wang et al., 2016; Bao and Xia,2017). Thus, based on RNA-Seq, we f irst identif ied potential diff erent functional genes participating in the transcriptomic responses ofM.nipponenseagainstA.veroniiorS.aureusinfection and provided insight into the diff erential response mechanism of crustaceans infected by gram-positive and gramnegative bacteria.
2 MATERIAL AND METHOD
2.1 Experimental animals and bacterial stimulation
Healthy juvenileM.nipponense(2-4 g per prawn)were obtained from a farm in Shaoguan, Guangdong Province, China. These oriental river prawns were acclimatized for one week in three aquariums (120 L)in the laboratory at room temperature, which was maintained at approximately 26 °C, and fed a commercial diet weighing 5% of the body weight at 18꞉00 once daily. First, a trial challenge of the prawns withA.veroniiorS.aureuswas conducted to determine the optimal concentration using three concentrations of bacterial cells (105, 106, and 107colony-forming unit (cfu)/mL) administered via second abdominal muscle injection; 25 μL of each concentration of bacterial cells was injected (20 prawns per group). After one week, the results showed that the mortality rate was more than 80% using 106and 107cfu/mL bacterial suspension with about 30%using 105cfu/mL. The infection and dissection experiments were all performed under MS-222 anaesthesia to minimize suff ering. The prawns in both the experimental and control groups were intramuscularly injected with equal volumes (25 μL)of a bacterial suspension at 105cfu/mL and phosphatebuff ered saline (PBS). According to the groups into an aquarium, the tested prawns were separately observed for mortality and sampled.
2.2 Sampling and RNA extraction
The prawns were sampled at 24-h post injection.Before sampling, twelve prawns from each group were anaesthetized with MS-222 and surgically dissected (including six female prawns and six male prawns). The hepatopancreatic tissue was sampled and immediately placed in RNA Keeper tissue stabilizer (Vazyme, China), stored at 4 °C for overnight and cryopreserved at -20 °C until RNA extraction.Total RNA extraction from the hepatopancreatic tissue was conducted by RNA Isolater (Vazyme, China)according to the manufacturer’s protocol. The integrity and quality of the total RNA were further measured using an Agilent 2100 bioanalyzer. RNA samples from two males and two females from each group were pooled as one biological duplicate for cDNA synthesis and sequencing.
2.3 Library construction and Illumina sequencing
After evaluation of RNA quality, a total of 1-μg RNA per sample was used for library preparation. The VAHTS mRNA-seq v2 Library Prep Kit for Illumina®(Vazyme, NR601) was further used to generate sequencing libraries following the manufacturer’s protocol. The following procedures, including RNA fragmentation, cDNA synthesis, size selection, PCR amplif ication and RNA-seq, were performed at Vazyme Biotech Co., Ltd. (Nanjing, China). In the typical procedure, mRNAs were fragmented into small pieces in Vazyme Frag/Prime buff er.Subsequently, random hexamer primers were used to synthesize the f irst-strand cDNA using Super Script II reverse transcriptase (Thermo Scientif ic, Delaware,USA) according to the manufacturer’s protocol.Using the QiaQuick PCR extraction kit (Qiagen,Germany), the short fragments were further purif ied to repair the end by adding a poly (A) tail. The library preparations were sequenced on an Illumina HiSeq X Ten platform with a 150-bp paired-end module.
2.4 Cleaning of raw data and de novo assembly
Before de novo assembly, the raw reads were trimmed by removing the dirty reads, which included reads with adaptors, low-quality sequences (reads with ambiguous ‘N’ bases at a ratio greater than 5%),and short-read-length sequences (in which the number of bases withQ≤10 was more than 50% of the entire read). Then they were further assembled into expressed sequence tag (EST) clusters (contigs) using the Trinity software with default parameters. The transcripts were further assembled and clustered by the Chrysalis clusters software with the default parameters. The longest assembled sequences obtained in each cluster were further designated as“unigenes”.
2.5 Functional unigene annotation and classif ication
The contigs that were assembled using the combined sequence data from the infected and noninfected samples were used as queries to search against three public databases by BLAST. All unigenes were further aligned against NCBI nonredundant protein database (Nr; http://www.ncbi.nlm.nih.gov/), the Kyoto Encyclopedia of Genes and Genomes (KEGG; http://www.genome.jp/kegg/), and the Clusters of Orthologous Groups of proteins database (COG; http://www.ncbi.nlm.nih.gov/COG/)with an E-value of 10-5using BLASTp (version 2.2.25). The BLAST results of the best hit of the unigenes were extracted for unigene description.Blast2GO was used to assign Gene Ontology (GO)terms to annotated contigs based on BLASTx hits against the NCBI Nr database (Ashburner et al.,2000). The unigenes associated with GO term were subsequently grouped into three categories including cellular component, biological process, and molecular function. The unigenes were also annotated against the COG database to identify the possible functional categories of the genes based on sequence similarities.Unigene information with respect to the KEGG pathways was obtained based on BLASTx hits against the KEGG database.
2.6 Diff erential gene expression analysis
All clean reads from each of the two groups(hepatopancreatic control (HC) vs. hepatopancreaticA.veronii(HA) and HC vs. hepatopancreaticS.aureus(HS)) were mapped to reference sequences(unigenes from the assembled transcriptome data)using Bowtie2 (Langmead and Salzberg, 2012). Gene expression levels were further calculated according to the fragments per kilobase per million mapped reads(FPKM). The diff erentially expressed genes (DEGs)between hepatopancreatic tissues of the two groups was identif ied using the R package edgeR (Robinson et al., 2010). To assess the signif icance of diff erential gene expression, the threshold for def ining signif icant DEGs was set as aP-value less than 0.05 and an absolute log2(fold change) value greater than 1. When the log2(fold change) was >1, the transcript was considered to be up-regulated, while the log2(foldchange) was <-1, the transcript was considered to be down-regulated. In addition, DEGs across the samples were further annotated by GO and KEGG pathway analysis. Only DEGs with annotation were considered candidates of interest for further analysis.
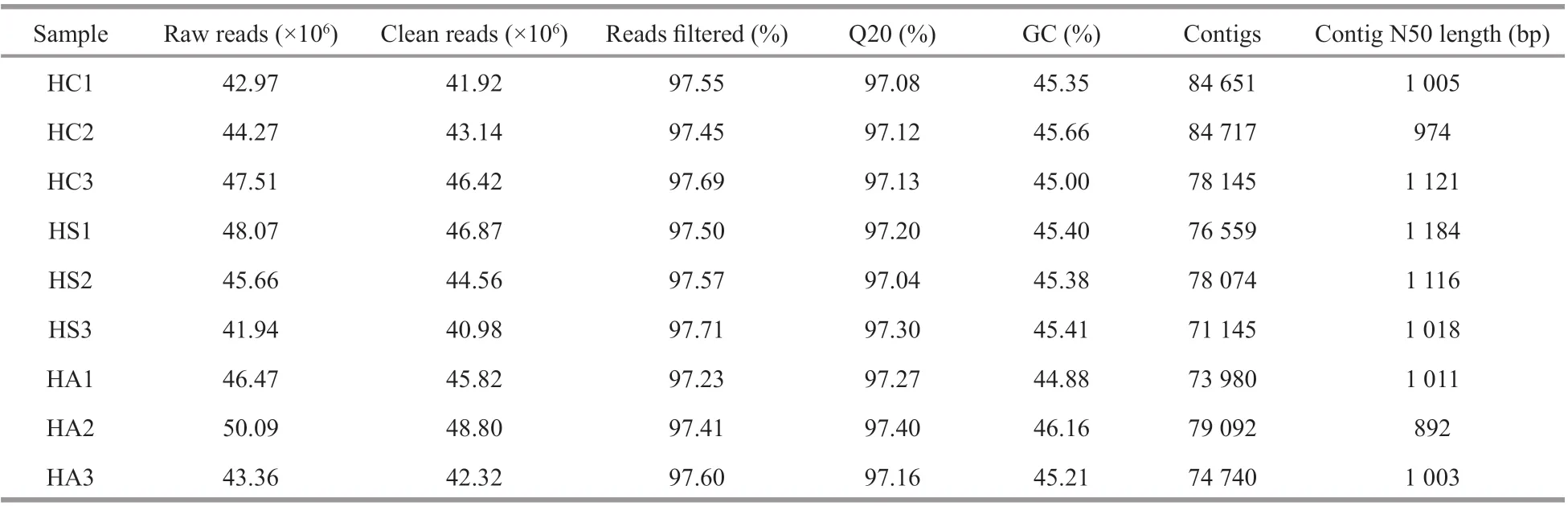
Table 1 Basic RNA-Seq data
2.7 Validation of diff erentially expressed genes using real-time PCR
To validate the reliability of the DEGs identif ied by RNA-Seq, we selected 15 genes involved in the immune system for quantitative Real-time polymerase chain reaction (qRT-PCR) validation. Based on each identif ied gene from the transcriptome library (Premier Biosoft, USA), PCR primers were designed using Primer Premier 6 software (Supplementary Table S1).qRT-PCR was performed using the AceQ qPCR SYBR Green master mix (Vazyme, Nanjing) by a LightCycle 480 system following the manufacturer’s instructions.The amplif ications were performed with the following program: 95 °C for 30 s, followed by 40 cycles of 94 °C for 15 s, 58 °C for 20 s, and 72 °C for 20 s. Target gene expression levels were normalized using β-actin and expressed as relative expression levels (relative mRNA expression). The dissolution curve was obtained at temperatures from 60.0 to 95.0 °C, increasing the temperature by 0.5 °C per 0.05 s. The actin gene was used as the reference gene (Xiu et al., 2013), and relative fold changes of DEGs were further calculated using the 2-ΔΔCtmethod (Livak and Schmittgen, 2001).
3 RESULT
3.1 Sequencing and de novo assembly
A total of 410.36 million raw reads were generated,including 134.75 million, 135.67 million, and 139.93 million reads from the hepatopancreatic tissue of the HC (HC1=42.97 million, HC2=44.27 million, and HC3=47.51 million), HS (HS1=48.06 million,HS2=45.66 million, and HS3=41.94 million), and HA(HA1=46.47 million, HA2=50.09 million, and HA3=43.36 million) groups, respectively (Table 1).After stringent quality assessment and data f iltering, a total of 400.19 million clean reads (97.52% of the total reads) were used for subsequent transcriptome assembly. The clean reads were assembled by Trinity after splicing and removing redundancy to create a total of 701 103 contigs. The Q20 value for each sample was more than 97%, and the N50 value was in the range of 892-1 184 bp, including 247 513 contigs in the HC group, 225 778 contigs in the HS group,and 227 812 contigs in the HA group. Most of the assembled unigenes (~69%) in each sample were shorter than 500 bp with the N50 value ranging from 1 371 to 1 743 bp (Table 2). When the reads of all the samples were merged for assembly, 56 944 unigenes were obtained with a total length of 71.34 Mbp. The unigenes shorter than 500 bp decreased to ~45% with the N50 value increase to 2 584 bp, indicating a marked increase in the quality.
3.2 Annotation of all unigenes
Unigene annotation provides information on the function of the unigene. First, using the BLAST tools,the 56 944 unigene sequences assembled from the combined reads were aligned to major databases,including the Nr, NT, Swiss-Prot, KEGG, COG, and GO databases (E-value<0.000 01). It was found that 21 754, 10 726, 18 595, 16 669, 9 907, and 11 578 unigenes had signif icant hits to these major databases,respectively. Totally, 24 688 unigenes were annotated by at least one database. According to the Nr annotation, approximately 46.6% of the unigenes had E-values less than 1e-45 and 61.5% shared greater than 40% similarity with known sequences (Fig.1). Interms of the species distribution of the most signif icant hits, 10.8% of the unigenes matched to sequences ofDaphnia pulex, which had the highest BLASTmatched ratio of the matched species.
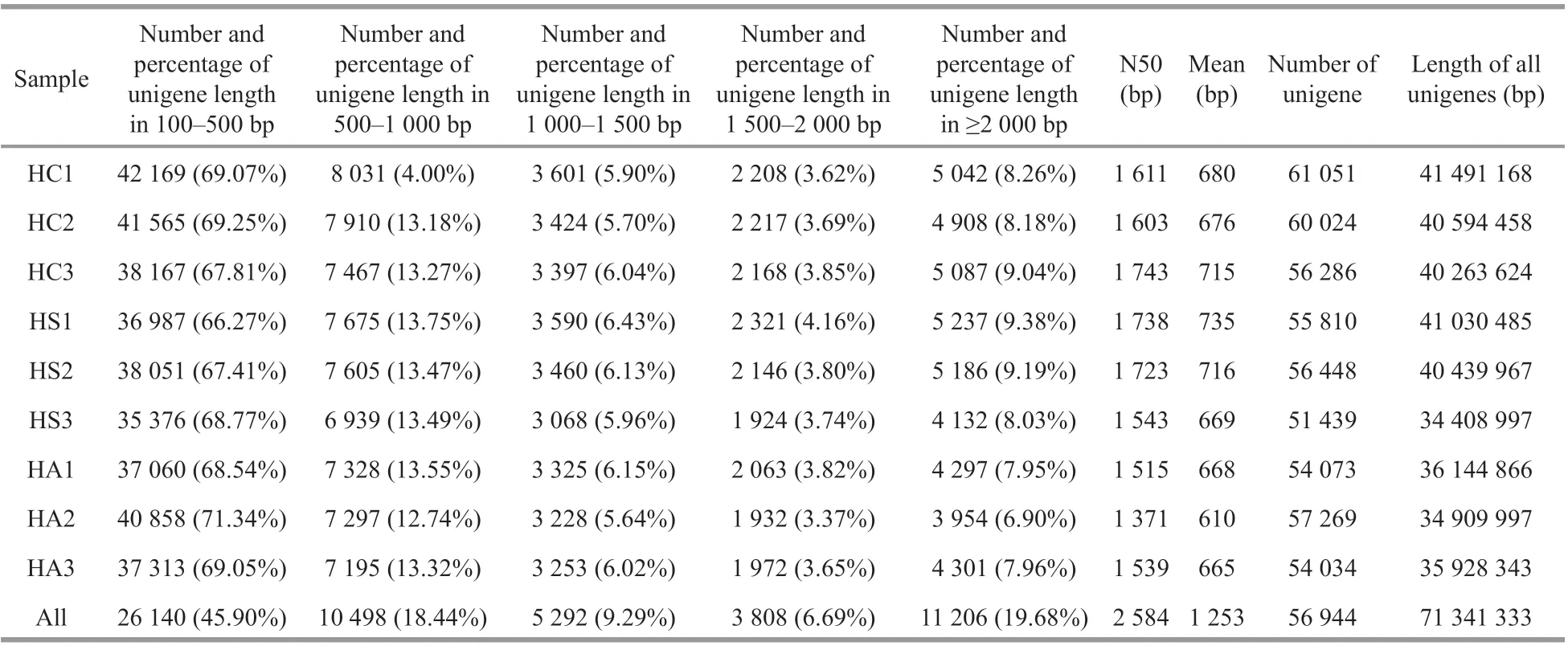
Table 2 Statistics of the unigene length distribution in each sample of the combined reads
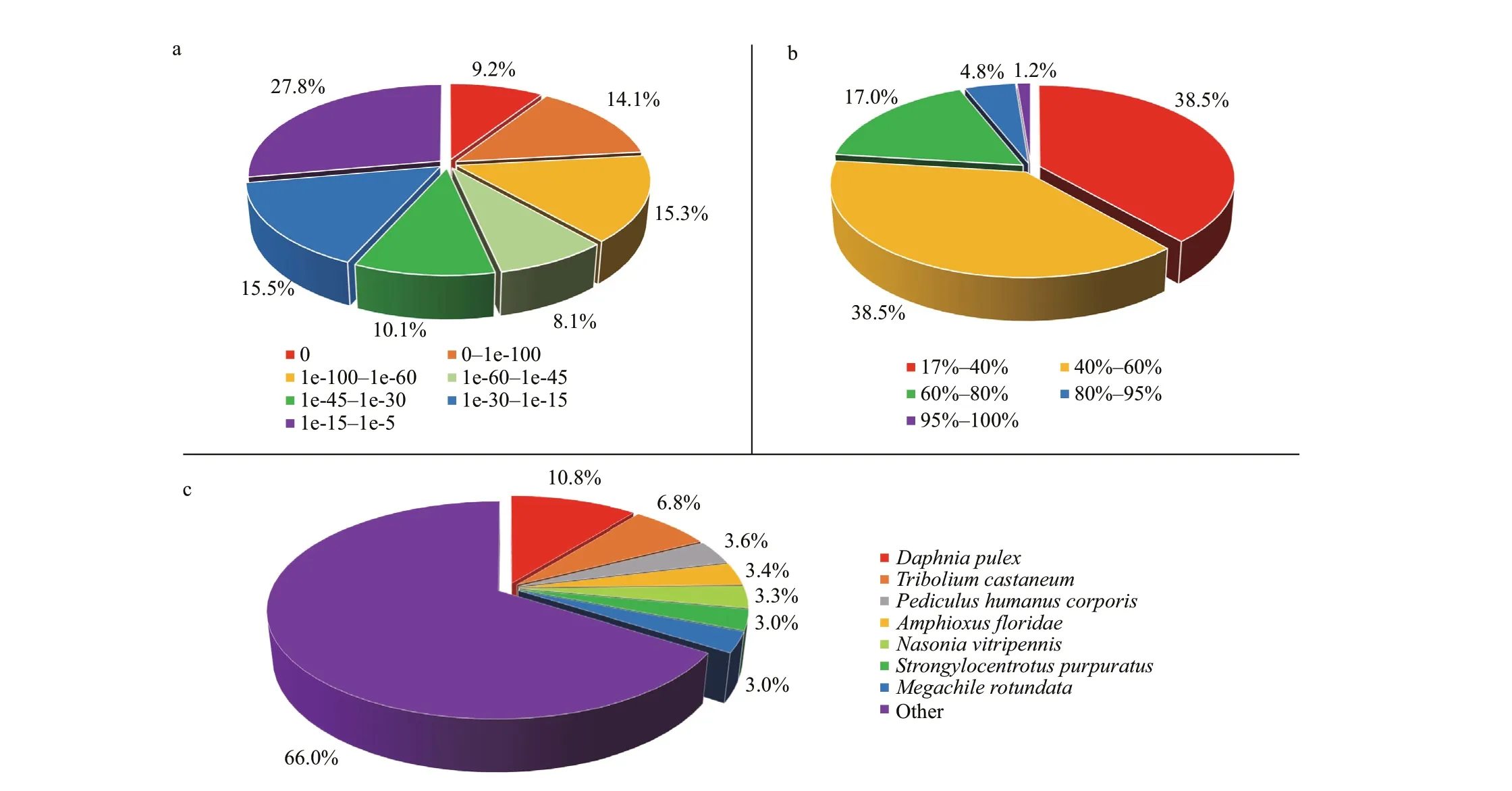
Fig.1 Summarized statistics of the Nr annotation
In the COG classif ication, 9 907 unigenes were categorized into 25 functional COG clusters. The top three enriched categories were general function prediction only (20.50%); replication, recombination and repair (8.19%); translation, ribosomal structure and biogenesis (7.67%) (Fig.2). In the biological process category of GO classif ication, the three most common subcategories were cellular process (7 927 unigenes), metabolic process (6 497 unigenes), and single-organism process (6 155 unigenes). Some immune-related GO terms were found in the biological process category, in which 2 887 unigenes were assigned to the term “response to stimulus”, followed by “immune system process” (551 unigenes) and“cell killing” (15 unigenes) (Fig.3).
KEGG pathway annotation was also conducted to study the associated biological pathways of the assembled genes. The results show that a total of16 669 unigenes were associated with 258 biological pathways in the KEGG database, such as metabolic pathways (2 404 genes), ubiquitin-mediated proteolysis (732 genes), regulation of the actin cytoskeleton (693 genes), and focal adhesion(556 genes). In addition, 15 pathways related to the immune system, mainly including the chemokine signalling pathway, leukocyte transendothelial migration, and the T-cell receptor signalling pathway,were further identif ied (Supplementary Table S2).
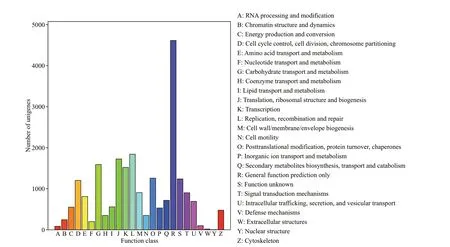
Fig.2 COG function classif ication of unigenes
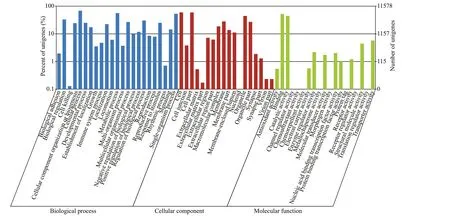
Fig.3 Summary of GO annotation
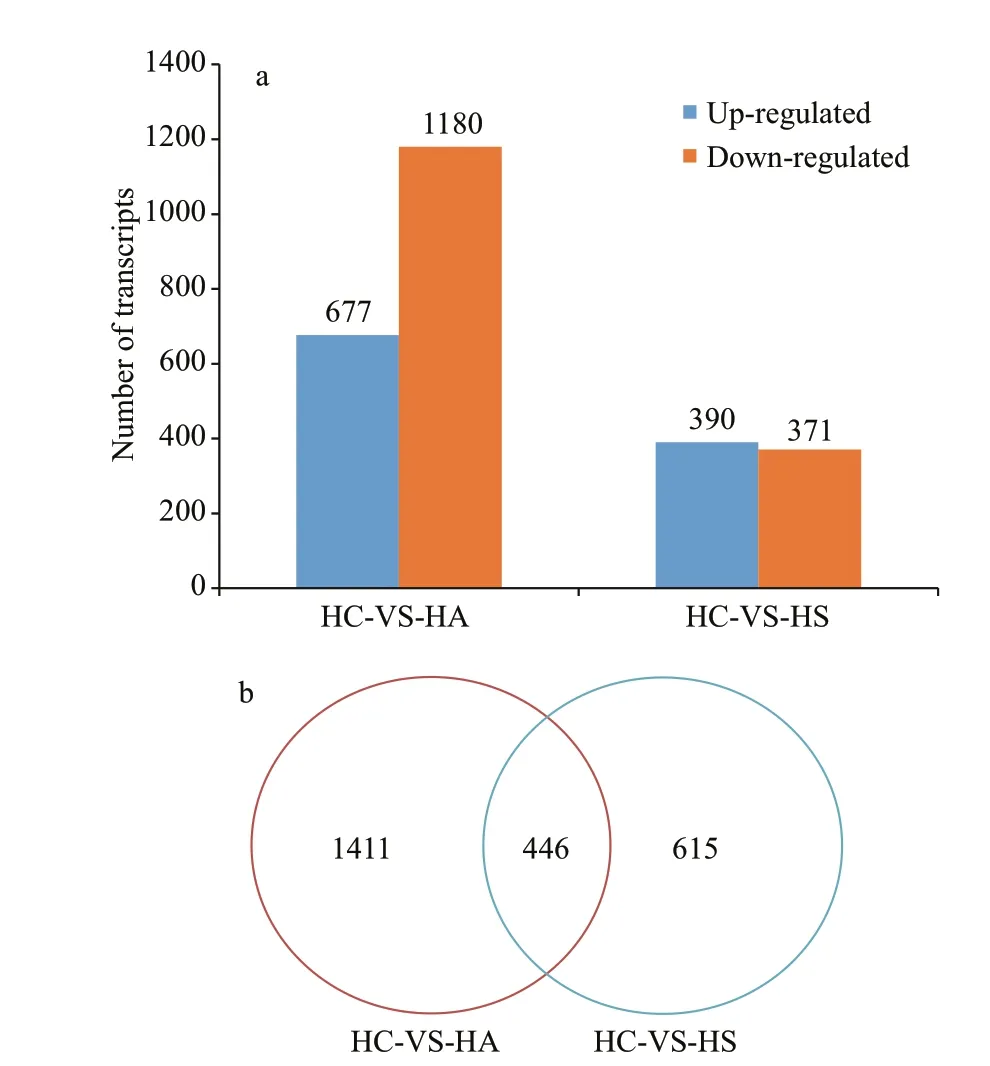
Fig.4 The diff erentially expressed genes were evaluated by bioinformatics analysis (a); venn diagram of diff erentially expressed genes (b)
3.3 Identif ication and annotation of diff erentially expressed genes
Comparison of gene expression levels in the HC vs. HA and HC vs. HS groups revealed 1 857 and 1 061 signif icant DEGs (|log2Ratio| ≥1, andP-value≤0.05), respectively. Among these DEGs, 677 genes were up-regulated and 1 180 were down-regulated in the HC vs. HA group (Supplementary Table S3;Fig.4a). A total of 390 genes were up-regulated and 671 were down-regulated in the HC vs. HS group(Supplementary Table S3; Fig.4a). About 446 DEGs were found in both HC vs. HA and HC vs. HS groups(Supplementary Table S3; Fig.4b).
To determine the biological functions of DEGs stimulated byA.veroniiorS.aureusinM.nipponense,GO classif ication and KEGG pathway enrichment analysis were performed. DuringA.veroniistimulation, GO functional analysis showed that the 287 signif icant DEGs could be annotated into 13 cellular components, 12 molecular functions, and 24 biological processes (Supplementary Table S4).Among the 24 categories of biological processes, the DEGs mostly participated in cellular process (189 genes), followed by the categories of single-organism process (183 genes). Within the 13 cellular component categories, the DEGs mostly participated in the cell(155 genes) and cell part (155 genes) categories. In the 12 molecular function categories, the DEGs mostly participated in the catalytic activity (146 genes)and binding (134 genes) categories. The KEGG pathway analysis annotated a total of 490 DEGs duringA.veroniistimulation, which were assigned to 220 KEGG pathways (Supplementary Table S5).Many immune-related pathways also exhibited signif icant unigene enrichment, including Pathways in cancer (81 genes), Natural killer cell mediated cytotoxicity (21 genes), Toll-like receptor signaling pathway (30 genes), and B cell receptor signaling pathway (15 genes) (Fig.5a).
After stimulation byS.aureus, the 189 signif icant DEGs could be annotated into 11 molecular functions,14 cellular components, and 25 biological processes by GO functional analysis (Supplementary Table S4).Among the 25 biological process categories, the DEGs mostly participated in the categories of cellular process(123 genes) and single-organism process (118 genes).Among the 14 cellular component categories, the DEGs were enriched in the cell (98 genes) and cell part (98 genes) categories. Within the 11 molecular function categories, the DEGs were annotated into the categories of catalytic activity (103 genes) and binding(85 genes). In addition, 42 DEGs were annotated in the response to stimulus, while 9 DEGs were annotated in the immune system process. A total of 312 DEGs duringS.aureusstimulation were annotated and assigned to 197 KEGG pathways (Supplementary Table S5). Metabolic pathways (46 genes) was the predominant category. Moreover, many immunerelated pathways were signif icantly enriched, including pathways in cancer (48 genes), B cell receptor signaling pathway (10 genes), and NOD-like receptor signaling pathway (17 genes) (Fig.5b).
3.4 Validation of diff erentially expressed genes
To validate the reliability of DEGs identif ied by RNA-Seq, some DEGs involved in the immune system were randomly selected for qRT-PCR validation. Levels of expression for the target genes isolated from hepatopancreatic tissue were determined in the infectedM.nipponenseand compared with those in the controls at 24 h post infection. These results are largely consistent with the transcriptomic prof ile analysis (Fig.6).
4 DISCUSSION
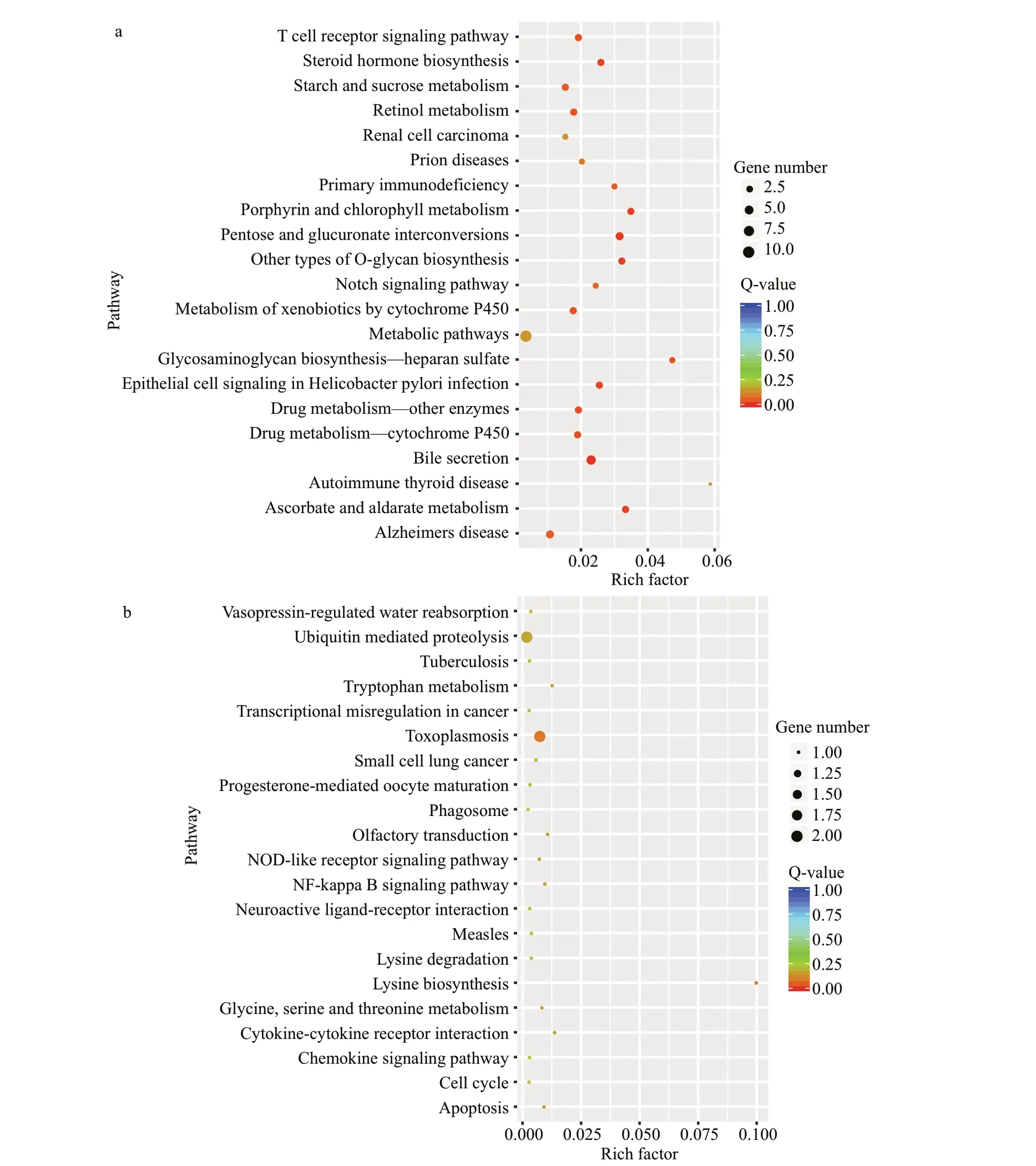
Fig.5 The statistics of KEGG pathway enrichment of diff erential expressed genes from HC-VS-HA (a) and HC-VS-HS (b)
Macrobrachium nipponenseis an economically important aquaculture species throughout China and in some other Asian countries. The aquaculture ofM.nipponensehas developed rapidly in recent years,but aquaculture environments with high stock density have experienced frequent disease breakouts.Comparative transcriptomic analysis has been widely used to understand the physiological response to various stimuli in crustaceans (Rao et al., 2015; Chen et al., 2017). In our study, comparative transcriptomic analysis was f irstly performed to assess the transcriptional response in the hepatopancreas ofM.nipponenseafter infection withA.veroniiorS.aureus. A total of 400.19 million clean reads were obtained and further assembled into 56 944 unigenes.Compared with the control samples (HC), 1857 DEGs were found in theA.veronii-infected (HA) samples and 1 061 DEGs were found in theS.aureus-infected(HS) samples. This research enriches theM.nipponensetranscriptome database and provides insight into the immune regulation of prawn in response to bacterial infection.
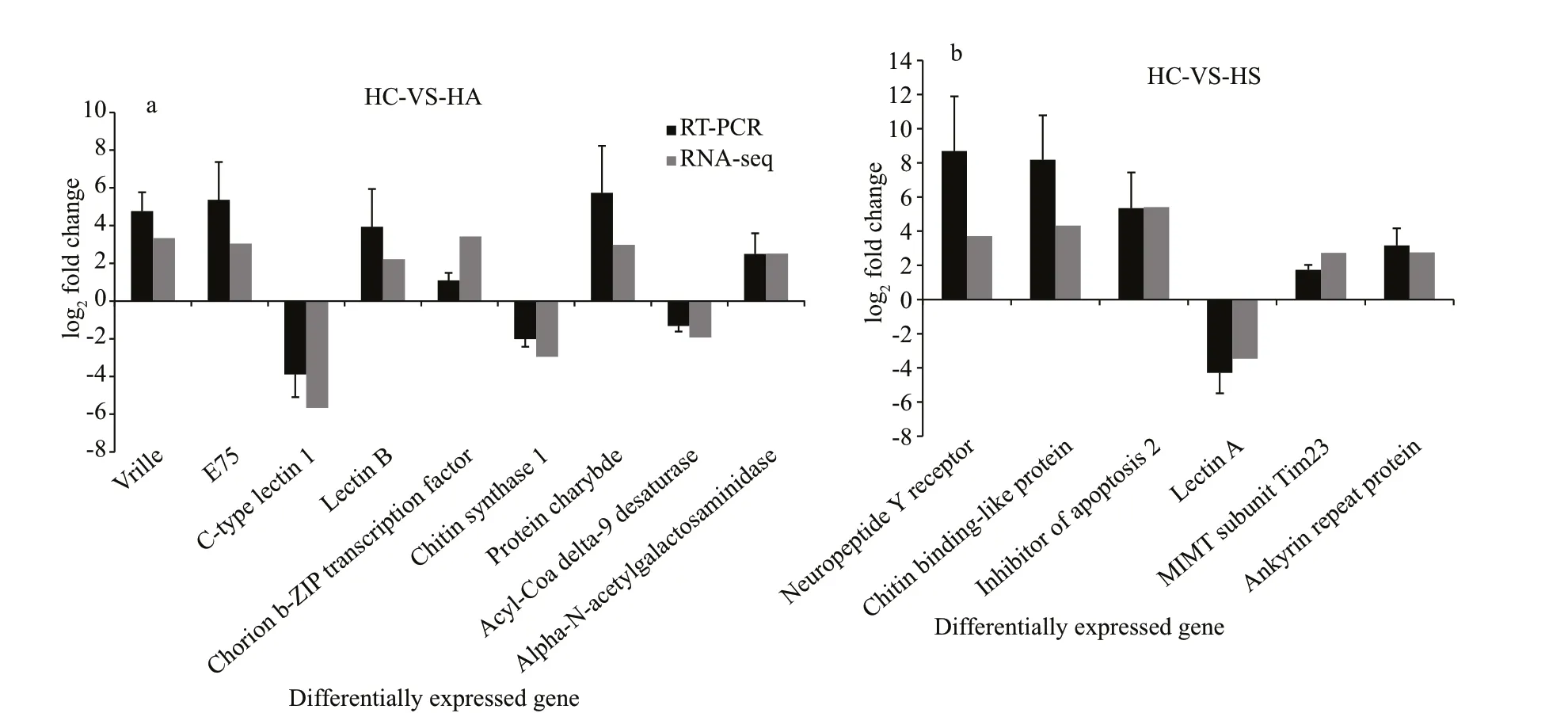
Fig.6 Comparison of the expression levels of transcriptome sequencing and qRT-PCR results
Immune-related DEGs are the target molecules involved in almost all transcriptomic analyses concerning the immune response. In fact, there were many immune-related DEGs potentially involved in responses toA.veroniiorS.aureusinfection,including prophenoloxidase (ProPO), mitochondrial manganese superoxide dismutase (mtMnSOD), tumor necrosis factor receptor-associated factor 6 (TRAF6),anti-lipopolysaccharide factor (ALF) and C-type lectin (Supplementary Table S3). Toll-like receptor signaling pathway plays an important role in the response to gram-negative and gram-positive bacteria by regulating a large number of genes (Pan et al.,2011). InDrosophila, the Toll pathway is triggered by spaetzle (Spz) cleavage, which starts an intracellular signaling cascade including DmMyD88, Tube(considered as the IRAK4 homologue), Pelle (the human IRAK1 homologue) and DmTRAF6, resulting in the translocation of Dorsal-related immunity factor or Dorsal (the homologue of human NF-kB) and the expression of Drosomycin (Belvin and Anderson,1996). As an extracellular ligand, the activated Spz binds as a dimer to the Toll ectodomain with high affi nity and with a stoichiometry of one Spz dimer to two receptor proteins: necrotic and persephone(Hoff mann, 2003; Weber, 2003). TRAF6, identif ied as a downstream target of Pelle, is a key signal adaptor that is involved in the interleukin-1 receptor/Toll-like receptor (IL-1/TLR) superfamily (Sun et al., 2017).Some studies have shown that TRAF6 acted as a bridge to link the upstream TLRs, IRAKs, and MyD88 with the NF-kB and MAPK signalling pathways(Arch et al., 1998; Kim and Rikihisa, 2002). In our data, Spz mRNA was up-regulated in the hepatopancreas after challenged withA.veroniiorS.aureus. Meanwhile, the TRAF6 was only up-regulated in theA.veronii-infected group. Shi et al. (2009) also demonstrated that the expression of theFcSpz transcript in the haemolymph, hepatopancreas and other tissues ofFenneropenaeus chinensiswas upregulated after injection withV.anguillarumand white spot syndrome virus. It has been reported that both Spz and TRAF6 could activate the promoters of certain antimicrobial peptide genes in vitro (Wang et al., 2007, 2011; Sun et al., 2017). In our transcriptomic data, ALF-3 that contains a conserved disulf ide loop and can neutralize lipopolysaccharide was signif icantly up-regulated afterA.veroniiorS.aureusinfection. Taken together, Spz and TRAF6 might play important roles in host defence against gram-positive and gram-negative bacteria invasion via regulating the expression of ALFs in crustaceans.
Most research attention on C-type lectin has focused on their roles in antimicrobial immunity.Classical C-type lectins contain carbohydrate recognition domains (CRDs) that bind carbohydrate structures in a Ca2+-dependent manner. Some C-type lectins are produced as transmembrane proteins, and others are secreted as soluble proteins (Cambi and Figdor, 2005). The C-type CRDs generate a subfamily including protein domains called C-type lectin-like domains (CTLDs). Some CTLDs can bind protein or lipid moieties, which are Ca2+independent (Zhang et al., 2009). C-type lectins have a binding site with affi nity for gram-negative bacteria, which have lipopolysaccharide (LPS) on their surface, and grampositive bacteria, which have other surface polysaccharides (Yu et al., 1999). In our study, several lectins, including C-type lectin and C-type lectin 1,were signif icantly up-regulated in the hepatopancreas ofM.nipponenseafter infection withA.veroniiorS.aureus. Similar expression of C-type lectins has also been observed inM.nipponenseinfected withAeromonashydrophilaorVibrio parahaemolyticus(Xiu et al., 2015b, 2016). In summary, these various types of lectins with structural and functional diversities were mainly expressed in the hepatopancreas of the prawn, which indicated that these proteins played essential roles in the defence system of the prawn.
ProPO is regarded as a critical part of the immune system and plays an important role in the immune recognition process in the defence mechanism of invertebrates. Upon injury or infection, the ProPO zymogen is activated to PO by clip-domain serine proteases, which are called PPAFs (Jiang et al., 1998;Satoh et al., 1999). PO compounds can produce quinones, which may kill pathogens and be used for synthesis of melanin to encapsulate parasites and seal wounds. In our transcriptome data, unigenes annotated as PPAF, serine proteinase homologue (SPH) and ProPO were exhibited signif icantly up-regulated afterA.veroniiinfection. Similar to this observation, inF.chinensis, bothFcSP andFcSPH can respond toV.anguillarumchallenge and displayed up-regulation(Ren et al., 2009). Serine proteases (SPs) and SPHs,which possess a trypsin-like domain at the C terminus and one or two cysteine-rich structural motif clip domains at the N terminus, are the essential components of extracellular signalling cascades in various biological processes of invertebrates (Lin et al., 2006; Jang et al., 2008). Furthermore, several groups have demonstrated that SPHs bind tightly to microbial cell wall components (Zhang et al., 2003)or pathogenic bacteria (Lee and Soderhall, 2001).Kan et al. (2008) determined thatTmSPH1 specif ically binds to curdlan polymers (β-1,3-glucan fungal polymer) and three components, namely, the serine proteaseTmSPE,TmproPO, andTmSPH1, and participates in melanin synthesis. Moreover, previous f indings demonstrated that one unique clip-domain serine protease (SPE) participated in the regulation of ProPO activation cascade and Toll signaling pathway(Kan et al., 2008). It is also noted that PO catalyses the production of quinones, which may be involved in the production of reactive oxygen species (ROS),such as superoxides and hydroxyl radicals.
ROS such as superoxides and hydroxyl radicals are constantly generated when the organism is attacked by pathogens, but overexpression of ROS will lead to cell damage. Therefore, a protective mechanism in the cell must exist to eliminate excess ROS. Cells are protected against oxidative stress by a series of antioxidant enzymes, such as superoxide dismutase(SOD), catalase (CAT), glutathione peroxidase(GPX), and peroxiredoxin (PRX). These antioxidant enzymes play important roles in sustaining homeostasis of the cell (Aruoma, 1998). In our transcriptomic data, antioxidant enzymes, thioredoxin(TRX), and mtMnSOD were up-regulated afterA.veroniichallenge, and in the HC vs. HS group,mtMnSOD was also signif icantly up-regulated.MtMnSOD can respond to various pathogenic invaders or foreign particles at the transcriptional level (Song et al., 2015). Meng et al. (2013)demonstrated that the expression level of the mtMnSOD gene was up-regulated inP.clarkiiafter challenge withSpiroplasma eriocheirisor
A.hydrophila. It was also demonstrated thatCfmtMnSOD mRNA transcripts could be signif icantly up-regulated by stimulation with three typical microbes in the hepatopancreas ofChlamys farreri(Wang et al., 2018). TRX is a major highly conserved and ubiquitous protein involving in protecting organisms against various oxidation pressures. Mu et al. (2009) reported that the expression of TRX in the hepatopancreas ofEriocheir sinensiswas upregulated after a gram-negative marine bacteriumA.veroniichallenge. Song et al. (2012) further demonstrated thatPtTRX1 transcripts are signif icantly up-regulated after challenged by the bacteriaV.alginolyticus, while the expression level ofPtTRX2 mRNA was up-regulated when injected with a grampositive bacteriaM.luteus. These results suggest that TRX might be more sensitive to gram-negative bacteria.
5 CONCLUSION
In conclusion, a comparative transcriptomic prof ile analysis of hepatopancreatic tissue fromM.nipponensestimulated byA.veroniiorS.aureuswas f irstly completed, and a large number of transcripts and DEGs involved in immune system were obtained. Although the functions of most genes and their associated pathways remain to be explored,this study provides much valuable data regarding the anti-bacterial immune mechanisms in prawn and f irst reveals a similar response mechanism of crustaceans after infection by gram-positive and gram-negative bacteria in transcriptome level. Furthermore, the transcriptomic analysis identif ied a large number of novel transcripts in the hepatopancreas ofM.nipponensethat were absent from the prawn genome database and will provide a stepping stone for further genomic studies on the molecular mechanisms associated with the late development of the hepatopancreas inM.nipponense.
6 DATA AVAILABILITY STATEMENT
All the raw data of nine sequencing libraries have been submitted to NCBI Sequence Read Archive with accession numbers SRR7665577, SRR7665578,SRR7665579, SRR7665580, SRR7665581,SRR7665582, SRR7665583, SRR7665584, and SRR7665585.
 Journal of Oceanology and Limnology2022年1期
Journal of Oceanology and Limnology2022年1期
- Journal of Oceanology and Limnology的其它文章
- Spatial diff erence in net growth rate of Yesso scallop Patinopecten yessoensis revealed by an aquaculture ecosystem model*
- Bacterial community composition in gut content and ambient sediment of two tropical wild sea cucumbers ( Holothuria atra and H. leucospilota)*
- Dietary intake of bamboo vinegar and charcoal powder (BVC)enhances resistance of African catf ish Clarias gariepinus to bacterial pathogen*
- Cloning of catalase gene and antioxidant genes in Scophthalmus maximus response to metalloprotease of Vibrio anguillarum stress*
- Taxonomy and regeneration of a newly recorded Polychaete Capitella teleta (Annelida, Capitellidae) in the coastal water of Shandong, China*
- Five new records of Xanthidae (Crustacea: Brachyura) from Hainan Island, China*