
丙烯腈/4-乙烯基吡啶辐射溶液共聚合
2022-03-05 11:11王雨松姜志文舒晶晶汪谟贞葛学武
辐射研究与辐射工艺学报 2022年1期
孙 锐 王雨松,2 姜志文 舒晶晶 汪谟贞 葛学武
1(中国科学院软物质化学重点实验室 中国科学技术大学高分子科学与工程系 合肥 230026)
2(中国科学技术大学合肥微尺度物质科学国家研究中心 合肥 230026)
聚丙烯腈(PAN)分子链上含有大量强极性的氰基,因而具有很好的耐化学试剂、耐酸碱性以及优异的力学性能,被广泛应用于制备多种纤维制品及膜材料[1-3]。PAN通常由丙烯腈(AN)自由基聚合得到,主要聚合方法有:均相溶液聚合、水相沉淀聚合、混合溶剂沉淀聚合、悬浮聚合和乳液聚合等[4-5]。均相溶液聚合是将AN(及共聚单体)和引发剂溶解在适当的溶剂中,在一定条件下引发单体的聚合反应,生成的PAN 溶于溶剂中,因此反应体系在聚合前后均为均相体系[6-7]。均相溶液聚合方法可以直接得到PAN 溶液,聚合工序简单,操作方便,得到的聚合液经脱单、脱泡之后可以直接用于纺丝、成膜等下步工序,因而成本较低,是PAN 工业生产中应用最多的一种聚合方式。AN 的均相溶液聚合常用的溶剂主要有:二甲亚砜(DMSO)[8-9]、N,N-二甲基甲酰胺(DMF)[10]和N,N-二甲基乙酰胺(DMAc)[11]。常用的引发剂是偶氮二异丁腈(AIBN)和偶氮二异庚腈(AIHN)。引发剂的片段残留在聚合物中,往往对聚合物的某些性能产生不良影响[12]。
此外,PAN 中的氰基虽然极性很高,但亲水性却很差,因而PAN存在着吸湿性差、回潮率低、易起静电、易被蛋白质污染等不足之处[13-17],这也制约了PAN 在纤维、超滤膜等领域的一些应用。因此,PAN 的亲水性改性已成为超滤膜材料等领域的一个研究热点。目前制备亲水性PAN 主要有两条途径:(1)在现有的PAN 基体上接枝亲水性高分子链[18]。Bagheri 等[19]使用AIBN 作为引发剂,通过自由基聚合在PAN 纤维上接枝聚甲基丙烯酸甲酯,提高了PAN 纤维的亲水性;陈志军等[20]以丙烯酰胺(AM)为接枝单体、过氧化二苯甲酰(BPO)为引发剂、三烯丙基异三聚氰酸酯(TAIC)为交联剂在丙酮水溶液中对PAN纤维进行了接枝亲水改性;(2)通过AN与亲水性单体的共聚合反应,直接在PAN 高分子链中引入亲水性官能团(如羟基、胺基、酰胺基和羧基等)[21-22]。如日本旭化成公司[23]采用AN 与丙烯酸、乙烯基吡啶和二羰基吡咯化合物等亲水性单体共聚,制备了亲水性良好的PAN纤维;刘晓辉等[24]采用自由基聚合方法,以AIBN为引发剂,甲基丙烯酸为共聚单体,DMF 为溶剂,进行了AN 的共聚合反应,制得亲水性聚丙烯腈;吴畅等[25]利用AIBN 作为引发剂,65oC下引发AN与4-乙烯基吡啶(4-VP)在甲苯中的自由基共聚反应,合成了亲水性的P(AN-co-4-VP)共聚物,再将共聚物溶解于DMF中,采用L-S 相转化法制备了具有抗污染性的P(AN-co-4-VP)超滤膜。化学法引发的AN 溶液共聚合反应的研究中也发现一些问题:例如残留的AIBN分解产物会对PAN的热性能产生影响;同时高温反应也会增加自由基向溶剂的链转移常数,使得聚合产物相对分子量不高。
γ射线辐射可以引发烯类单体进行均聚及共聚反应。与传统的化学引发聚合方式相比,辐射引发聚合方式的优点在于:反应体系无需加入化学引发剂,产物纯度高,且无需加热,聚合反应可以在室温或更低的温度下进行[26]。葛学武等[27]采用60Co γ 射线辐射引发丙烯腈-丙烯酸甲酯-衣康酸(AN-MA-IA)三元体系在水-DMF混合溶剂中进行沉淀共聚合的方法,成功合成了重均相对分子量≥5×105的AN-MA-IA共聚物。赵新等[28]用核磁共振(NMR)谱表征方法对丙烯腈均相辐射聚合机理进行了初步的讨论。发现当吸收剂量和剂量率较大(50 Gy/min)时,聚合链末端单体联结方式对单体接到链上的概率无影响,即单体的加成服从Bernouilan过程,生成多种立构结构,说明该条件下丙烯腈的辐射聚合符合自由基聚合机理。但辐射引发AN均相溶液(共)聚合的工作却几乎未见报道,一个可能的原因是溶剂的辐射化学效应复杂,容易引起链终止或链转移,导致聚合产物相对分子量不高,性能较差。
本文以AN与4-VP的DMSO溶液为研究对象,开展了γ射线辐射溶液共聚合制备AN/4-VP二元共聚物的研究。考察了二元共聚反应的总单体转化率和共聚物溶液黏度随剂量率和吸收剂量的变化规律;并通过红外光谱、核磁共振波谱、热重分析仪、差示扫描量热分析仪和接触角仪等对所合成的二元共聚物的化学结构、热性能和亲水性进行了表征分析。
1 材料与方法
1.1 原料与试剂
AN、4-VP和DMSO均为分析纯试剂,购于国药集团化学试剂有限公司。4-VP经碱性Al2O3层析柱纯化后,置于冰箱(-20oC)中保存备用。其他试剂未经纯化,直接使用。液相色谱级DMF 购于上海阿拉丁生化科技股份有限公司。所有实验用水均为去离子水。
1.2 AN/4-VP辐射溶液共聚合
称取一定质量的AN 和4-VP 溶解于DMSO 溶液中,配置成不同单体质量分数的溶液。将适量单体溶液倒入辐照瓶中,连续通入氮气10 min(30 气泡/min)后,盖上瓶塞,并用Parafilm M 封口膜密封,再放入60Co源室中,在一定剂量率下室温辐照特定时间,得到聚合物溶液。60Co γ射线放射源位于中国科学技术大学,活度为7.4×1014Bq,剂量率采用丙氨酸/EPR标准剂量计标定。
1.3 表征方法
1.3.1产率和单体转化率的测定
取两块干净的自制方玻璃板,总质量记为m0。将适量聚合物溶液滴在其中一块上,再盖上另一块,用力将玻璃板之间的聚合物溶液压成薄膜状。玻璃板和薄膜的总重记为m1。参考文献[29]中聚丙烯腈沉淀分离的方法,将夹着聚合物溶液的两块玻璃板再分开,同时依次浸入75%、40%和20% DMSO 水溶液中,每种沉淀液中各浸泡30 min。接着用去离子水再浸泡30 min,并使凝固的聚合物薄膜从玻璃板上分离,最后将聚合物薄膜用丙酮浸泡30 min,放入在60 ℃真空烘箱内烘干至恒重。将玻璃板和干燥的聚合物薄膜一起称重,总质量记为m2。根据公式(1)计算得到聚合物的产率(Y,%)。

式中:C0为聚合前单体溶液中总单体质量分数。
因辐射引发丙烯腈聚合属于链式聚合机理,单体一旦引发即迅速生成高分子链,因此总单体转化率(简称单体转化率)可认为近似等于产率[30]。
本研究其他实验中所用的干燥聚合产物样品均用以上沉淀法分离得到。
1.3.2聚合物溶液黏度的测定
取少量聚合物溶液,用SNB-2 型旋转黏度计(4号转子)测定溶液的动力黏度。
1.3.3聚合物相对分子量的测定
(1)凝胶渗透色谱(GPC)法。称取约5 mg干燥聚合产物溶于1 mL HPLC 级DMF 中。取0.5 mL 溶液经0.22 μm 过滤器(NY 有机系针头过滤器,13 mm×0.22 μm)过滤后,注入Waters 1515型凝胶色谱仪中进行测试。使用的色谱柱为聚苯乙烯凝胶,检测器型号为Waters 2414,标样为聚苯乙烯,测试温度为40 ℃,流动相为DMF,流速为1.0 mL/min。
(2)黏度法。称取80 mg 干燥聚合产物加入25 mL DMF 中。室温下搅拌24 h,使聚合物充分溶解。移取10 mL 聚合物溶液注入放置在(30±0.1)℃水浴中的乌氏黏度计(φ0.55 mm)中。恒温10 min左右后,测定聚合物溶液的流出时间(t)以及纯DMF 溶剂的流出时间(t0)。每个样品的流出时间都测3 次以上,取平均值,代入公式(2)和(3)中,计算测试溶液的相对黏度(ηr)和增比黏度(ηsp)。

以溶液质量浓度(C)为横坐标,ηsp/C、lnηr/C分别为纵坐标作图,得到两条直线,将其外推于C=0 处,求出聚合物溶液的特性粘数[η]。再按公式(4)[27,31]计算得到共 聚物的重均相对分 子量(MW)。

1.3.4红外光谱测定
聚合产物的傅里叶变换红外光谱(FTIR)采用KBr 压片法在Nicolet 6700 红外光谱仪上测得。测量波数范围为4 000~400 cm-1,仪器分辨率为4 cm-1,扫描次数16次。
1.3.5核磁共振波谱测定
聚合产物的1H-NMR 和13C-NMR谱图由Bruker AVANCE AV III 400 型核磁共振波谱仪测得,溶剂为DMSO-d6,四甲基硅烷(TMS)作内标。13C 核的观察频率为100 MHz,脉冲序列为反转门控去耦zgig(Bruker 标准脉冲序列),测试温度60 ℃,脉冲延迟6 s,采样次数大于3 000次。
1.3.6热失重分析(TGA)
取干燥聚合产物4~6 mg,用热重分析仪(TGA 50H)测定样品的热失重曲线。样品在空气气氛中以10 °C/min 的升温速率,从25 ℃加热至850 ℃。
1.3.7差示扫描量热(DSC)分析
取干燥聚合产物2~4 mg,用差示扫描量热分析仪(DSC 60)记录样品的DSC 曲线。样品在氮气气氛中加热,升温速率为5 ℃/min,扫描温度范围为25~350 ℃。
1.3.8聚合物膜静态水接触角测试
将干燥的聚合物膜放在载玻片上,用接触角测定仪(SL200B,Solon Tech.Co.,Ltd.)测量其静态水接触角,水滴体积为2µL。
2 结果与讨论
高能射线穿过单体溶液时,可以同时引起单体分子和溶剂分子的辐射分解,生成各种活泼的离子或自由基,引发单体的聚合反应。因此辐射溶液聚合过程与单体和溶剂分子的化学结构、单体含量、吸收剂量和剂量率等因素密切相关。本工作中,我们选定AN 和4-VP 为共聚单体、相对物质的量比(nAN∶n4-vp)为98∶2、共聚单体总质量分数为20%的AN/4-VP 的DMSO 溶液体系为研究对象,首先考察了剂量率对AN/4-VP 溶液共聚合过程的影响。
2.1 剂量率对单体转化率和聚合物溶液黏度的影响
图1 列出了在不同剂量率下,吸收剂量为30 kGy 时测得聚合反应体系的单体转化率和聚合物溶液黏度。数据表明,剂量率在8~66 Gy/min内,单体转化率和聚合物溶液黏度均与剂量率相关。剂量率低于11 Gy/min时,单体转化率随剂量率增大而增加。当剂量率为11 Gy/min时,转化率可达94%。与此同时,所得聚合物溶液的黏度较高(大于250 Pa·s),并且也随着剂量率增大而增加。这是因为在较低剂量率范围内,随着剂量率的增加,单位时间内产生的自由基增多,单体的聚合速率增加,转化率上升,使所得聚合物浓度增加,进而使溶液黏度增大。
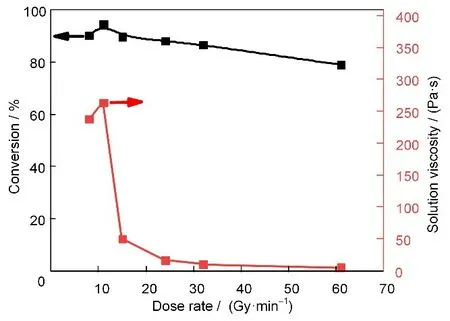
图1 在不同剂量率下,当吸收剂量为30 kGy时,AN/4-VP辐射溶液共聚合体系的单体转化率和聚合物溶液黏度(溶剂:DMSO;总单体质量分数:20%;nAN:n4-VP=98:2)Fig.1 The monomer conversion and viscosity of polymer solution measured after the DMSO solution of AN/4-VP was irradiated by γ-ray radiation at different dose rates.(Total absorbed dose:30 kGy;Monomer mass fraction:20%;nAN:n4-VP=98:2)
但剂量率超过11 Gy/min 后,在相同的吸收剂量下,单体转化率却随着剂量率的增加而缓慢下降至80%。值得注意的是,所得聚合物溶液的黏度骤降至20 Pa·s 以下。聚合物溶液的黏度不仅与聚合物浓度相关,还与聚合物相对分子量大小相关[32]。在较高剂量率下,聚合体系单体转化率仍保持在80%~90%,说明聚合物浓度应该变化不大,这意味着黏度的骤降应为所得聚合物相对分子量下降导致。这说明剂量率过大时,虽然生成的活性中心浓度增加、但同时也导致链终止速率增加,最终使得聚合物的相对分子量下降,溶液黏度降低。相对分子量与剂量率的这种依赖关系也出现在AN 的辐射本体聚合中[28]。根据以上实验结果,我们选定剂量率为11 Gy/min,进一步研究AN/4-VP 辐射溶液共聚合体系的单体转化率和聚合物溶液黏度随吸收剂量的变化关系。
2.2 单体转化率和聚合物溶液黏度随吸收剂量的变化
图2 列出了剂量率为11 Gy/min 时,不同吸收剂量下测得的单体转化率和聚合物溶液黏度。当吸收剂量从11 kGy 增加至27 kGy 时,单体转化率随着吸收剂量而增加,从78%上升至92%,这与辐射自由基聚合机理相符。此后,即使吸收剂量继续增加转化率也几乎保持不变,说明吸收剂量为27 kGy时,单体基本已反应完全。
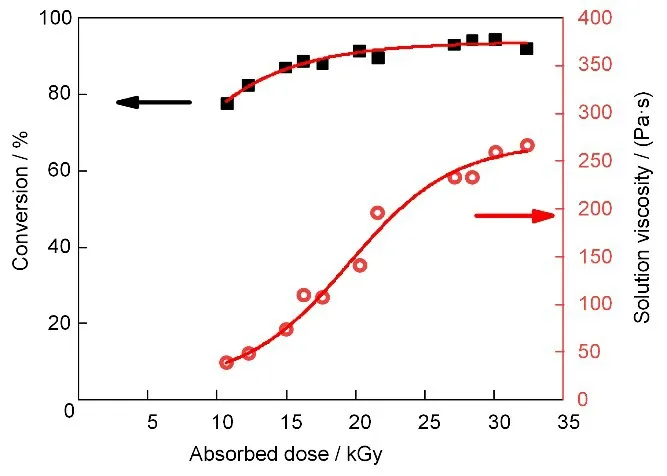
图2 剂量率为11 Gy/min时,AN/4-VP辐射溶液共聚合体系单体转化率和聚合溶液黏度随吸收剂量的变化(溶剂:DMSO;总单体质量分数:20%;nAN:n4-VP=98:2)Fig.2 Effect of the absorbed dose on the monomer conversion and the viscosity of polymer solution in the γ-ray radiation induced copolymerization of AN and 4-VP in DMSO.(Dose rate:11 Gy/min;monomer mass fraction:20%;nAN:n4-VP=98:2)
与此同时,所得聚合物溶液的黏度在所考察条件下却随着吸收剂量的增加一直在上升。通常在同一种溶剂中,聚合物浓度越高,相对分子量越大,其溶液黏度也越高。因此,随着转化率升高,聚合物浓度增加,从而使聚合物溶液黏度增加。但当吸收剂量大于27 kGy 后,转化率基本保持不变,即聚合物浓度基本不变,此时聚合物溶液黏度随吸收剂量的增加意味着体系中聚合物的相对分子量在继续增大。这体现了辐射引发聚合与化学引发聚合的不同之处,因为在聚合过程中高能射线同时作用于已生成的高分子链,在其上产生活性自由基。当高分子链的浓度越来越高时,高能射线在高分子链上产生的自由基之间相互接触的概率增大,最终有可能相互结合形成更高相对分子量的高分子链。极端情况下,即当吸收剂量足够高时,溶液中的高分子链相互之间形成交联网络,导致体系黏度剧增。PAN 的辐射交联效应已有很多报道[33-34]。在本工作中,当吸收剂量为30 kGy 时,转化率为94%,聚合溶液黏度为260 Pa·s;GPC 测得的共聚物重均相对分子量为1.3×105;而当吸收剂量达到60 kGy 时,整个体系成为凝胶状(图3)。

图3 吸收剂量为30 kGy和60 kGy时得到的辐射溶液聚合体系外观(溶剂:DMSO;总单体质量分数:20%;nAN:n4-VP=98:2;剂量率:11 Gy/min);内插图为吸收剂量为30 kGy时所得聚合物的GPC曲线Fig.3 Appearance of DMSO solution of AN and 4-VP after being irradiated by γ-ray radiation at an absorbed dose of 30 kGy and 60 kGy,respectively(Monomer mass fraction:20%;nAN:n4-VP=98:2;dose rate:11 Gy/min);the inset is the GPC curve of the polymer synthesized at an absorbed dose of 30 kGy
对剂量率为11 Gy/min、吸收剂量为30 kGy辐照条件下所得的PAN 均聚物及P(AN-co-4-VP)二元共聚物进行了一系列表征和对比分析。P(AN-co-4-VP)二元共聚物与PAN 均聚物的FTIR光谱如图4(a)所示。两者的IR 光谱上都出现了对应于游离羟基的伸缩振动(3 500~3 700 cm-1)和弯曲振动峰(1 630 cm-1),这可归因于样品在DMSO 水溶液凝固浴中沉淀析出过程中吸附的微量水。2 940 cm-1和2 870 cm-1的吸收峰归因于亚甲基C-H 的伸缩振动,2 244 cm-1处的吸收峰为氰基(-C≡N)的特征吸收峰,1 457 cm-1处的吸收峰为亚甲基C-H的弯曲振动峰,1 357 cm-1和1 250 cm-1处的吸收峰分别为次甲基C-H 的弯曲振动峰和摇摆振动峰,1 072 cm-1处的吸收峰为C-C 的伸缩振动峰,540 cm-1处的吸收峰对应于与氰基相连的CC(≡N)的弯曲振动[35]。与PAN的IR光谱相比,P(AN-co-4-VP)的IR光谱上出现了明显的吡啶环的特征吸收峰:1 597 cm-1、1 555 cm-1和1 414 cm-1处为吡啶环骨架的伸缩振动吸收峰,1 000 cm-1处为C-H面内弯曲振动峰,825 cm-1处为C-H面外弯曲振动峰。
P(AN-co-4-VP)二元共聚物和PAN 均聚物的1H NMR 谱图如图4(b)所示,其中δ=1.75~2.30 为高分子链中-CH2-单元中H 原子的化学位移;δ=3.00~3.25高分子链中-CH-单元中H原子化学位移;δ=2.5为DMSO-d6中H原子化学位移;δ=3.3 为体系吸收的水峰。在δ=8.57 和δ=7.35 处的两个峰分别为吡啶环上与N相邻和相间的C上所连H原子的化学位移。将δ=7.35 的核磁共振峰面积定为1,共聚物主链上所有-CH2-单元H 原子的共振峰的相对峰面积则为44.79,由此计算出共聚物分子链上4-VP 单元的物质的量百分数为(1/44.79)×100%,即2.2%[36],这与共聚单体投料物质的量百分数(2%)相当,这也说明4-VP几乎全部参与了共聚反应。
13C NMR谱具有宽的化学位移范围,能够区别分子结构中化学环境差异微小的碳原子,而且可以观察到不与氢核直接相连的含碳官能团,能够提供比1H NMR 谱图更多的碳骨架结构信息。P(AN-co-4-VP)二元共聚物和PAN 均聚物的13CNMR 谱图如图4(c)所示。其中δ=27.2~28.4 为PAN中-CH-单元中C原子的核磁共振峰,δ=32.9~33.3为-CH2-单元中C原子的核磁共振峰,δ=39.4~40.6为溶剂DMSO-d6中C原子的共振峰,δ=120.0~120.9为-C≡N上C原子的共振峰。在δ=121~160的放大图中可以清晰看到,P(AN-co-4-VP)二元共聚物谱图中出现了吡啶环上两组C原子的核磁共振峰δ=150.5和δ=123.7,这与1H NMR谱的表征结果一致。
PAN 分子链中次甲基(-CH-)碳为不对称碳原子,根据其所连氰基的空间排列方式(图4(c)),PAN 分子链的空间立构可分为全同立构(mm)、间规立构(rr)和无规立构(mr)。这3种空间构型所对应的次甲基C原子在核磁共振谱中的化学位移并不相同,因而可以利用PAN13C NMR谱图中次甲基碳的这3个不同化学位移峰的积分面积比表征所合成的PAN 的等规度(定义为:全同立构和间规立构聚合物在聚合物总量中所占的百分数,即((mm+rr)/(mm+rr+rm))×100%。小于50%为无规立构聚合物)[6]。从图4(c)中-CH-单元上C 的核磁共振峰放大图(δ=27.2~28.4)可清晰看出包含3 个峰,与文献中研究结果[28,37]对比分析确定,δ=27.20、δ=27.88 和δ=28.36 分别可归属为mm、mr 和rr 三单元组中次甲基碳的化学位移。通过3 个峰的积分面积,可以计算得到P(AN-co-4-VP)二元共聚物和PAN均聚物的等规度分别为48.6%和48.7%,均小于50%。这说明辐射溶液聚合得到的P(AN-co-4-VP)共聚物和PAN均为无规立构分布,4-VP 结构单元的引入并不改变PAN聚合物的等规度。
P(AN-co-4-VP)二元共聚物和PAN均聚物的DSC 曲线如图4(d)所示。少量吡啶的引入对PAN的热转变行为几乎没有影响。二者均在270 ℃有一个尖锐的放热峰。众所周知,PAN 分子链中的-C≡N侧基在高温时容易发生环化反应,放出大量的热[38-39]。本课题组之前的工作[40]以及大量文献研究[9,41-43]均表明,在N2气氛中,PAN 发生环化反应最大的放热峰对应的温度范围为270~300 ℃。这说明辐射溶液聚合得到的PAN 和P(AN-co-4-VP)二元共聚物与化学法合成的产物的热效应相似。
图4(e)P(AN-co-4-VP)二元共聚物和PAN均聚物在空气气氛中的TG曲线表明二者的热分解过程也基本相同,除了在380~600 ℃,P(AN-co-4-VP)二元共聚物的热失重比例比PAN多出4%左右。对于PAN 均聚物来说,在空气气氛下380~600 ℃的热失重主要来自伴随着分子内环化或分子间交联以及氧化、脱氢等反应过程中释放出多种小分子气体,以及没有发生环化或交联的大分子链的热裂解形成的小分子气体逸出[41]。而根据图4(b)中1H NMR 的表征结果,P(AN-co-4-VP)二元共聚物中4-VP的质量含量恰好约为4%,这说明4-VP结构单元的在加热过程可能没有参与-C≡N侧基的成环或交联反应,而是直接发生热降解,因为均聚的聚4-乙烯基吡啶在450oC 左右100%热解[44-45]。

图4 辐射溶液聚合所得PAN均聚物和P(AN-co-4-VP)二元共聚物的FTIR光谱(a);1H NMR谱图(b);13C NMR谱图(c);DSC谱图(d)和TG曲线(e)(溶剂:DMSO;剂量率:11 Gy/min;吸收剂量:30 kGy;总单体质量分数:20%;nAN:n4-VP=98:2)Fig.4 FTIR(a),1H NMR(b),13C NMR(c)spectra,DSC(d)and TG(e)curves of PAN and P(AN-co-4-VP)copolymer synthesized by radiation-induced solution polymerization(Solvent:DMSO;dose rate:11 Gy/min;absorbed dose:30 kGy;monomer mass fraction:20%;nAN:n4-VP=98:2)
2.3 总单体质量分数对单体转化率和聚合物溶液黏度的影响
在剂量率为11 Gy/min、吸收剂量为30 kGy且保持AN 和4-VP 相对比为98∶2 的条件下,聚合体系的单体转化率和所得聚合物溶液黏度随总单体质量分数的变化如图5 所示。在相同的辐照条件下,单体转化率基本与单体质量分数无关,均在95%左右。但聚合物溶液的黏度随着总单体质量分数而增加,单体质量分数低于15%时,聚合物溶液黏度很低,不超过1 Pa·s;单体质量分数继续增加,聚合物溶液的黏度迅速增大。单体质量分数为20%时,溶液黏度已达260 Pa·s,单体质量分数增至30%时,溶液黏度超过了6 000 Pa.s,室温下几乎无流动性。这与辐射自由基聚合体系中单体转化率和聚合物溶液黏度与单体质量分数之间的变化关系一致[46-47]。转化率相同的情况下,总单体质量分数增加时,所得聚合物溶液的浓度显然也增大,同时活性链有效碰撞概率增大,使聚合物链之间的化学键接概率也大大增加,形成交联网络的速度也增加,因而导致体系黏度迅速增大。
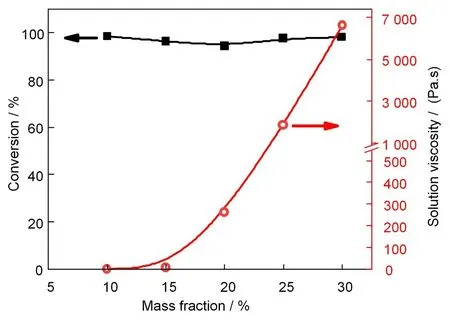
图5 不同单体质量分数下,AN/4-VP辐射溶液共聚体系的单体转化率和所得溶液黏度(溶剂:DMSO;剂量率:11 Gy/min;吸收剂量:30 kGy;nAN:n4-VP=98:2)Fig.5 Effects of the monomer mass fraction on the monomer conversion and the viscosity of polymer solution in the γ-ray radiation induced copolymerization of AN and 4-VP in DMSO(Dose rate:11 Gy/min;absorbed dose:30 kGy;nAN:n4-VP=98:2)
2.4 共聚单体相对物质的量比对单体转化率、溶液黏度以及共聚物亲水性的影响
在剂量率为11 Gy/min、吸收剂量为30 kGy且保持总单体质量分数为20%条件下,聚合体系的单体转化率和所得聚合物溶液黏度随AN 和4-VP相对物质的量比的变化如图6 所示。由图6,辐照条件和总单体质量分数均相同,且4-VP 共聚单体所占比例在不超过10%的范围内,辐射溶液聚合的单体转化率与4-VP 含量几乎无关,都保持在90%左右,但所得聚合物溶液的黏度却随着4-VP相对物质的量含量的增加迅速降低。因此我们用黏度法对图6 中各聚合产物进行了相对分子量测定,结果如图7所示。
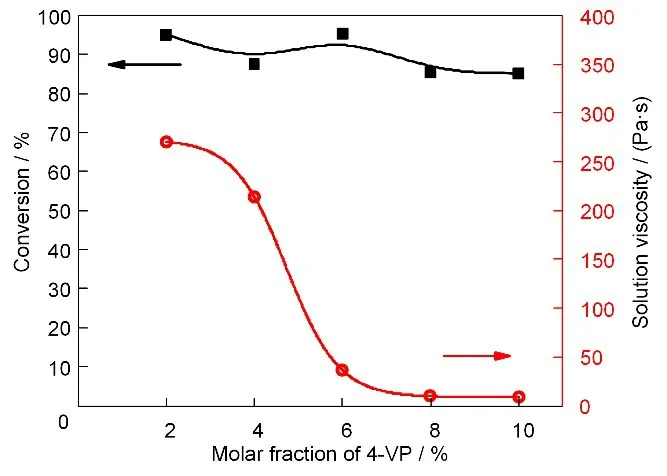
图6 AN/4-VP辐射溶液共聚合体系单体转化率和聚合物溶液黏度随共聚单体相对物质的量比的变化(溶剂:DMSO;剂量率:11 Gy/min;吸收剂量:30 kGy;总单体质量分数:20%)Fig.6 Effects of the molar fraction of 4-VP on the monomer conversion and the viscosity of polymer solution in the γ-ray radiation induced copolymerization of AN and 4-VP in DMSO(Dose rate:11 Gy/min;absorbed dose:30 kGy;monomer mass fraction:20%)

图7 AN/4-VP辐射溶液共聚合所得聚合物相对分子量随共聚单体相对物质的量比的变化(溶剂:DMSO;剂量率:11 Gy/min;吸收剂量:30 kGy;总单体质量分数:20%)Fig.7 Effects of the molar fraction of 4-VP on the molecular weight of P(AN-co-4-VP)prepared by the γ-ray radiation induced copolymerization of AN and 4-VP in DMSO(Dose rate:11 Gy/min;absorbed dose:30 kGy;monomer mass fraction:20%)
图7的结果表明,共聚物的相对分子量明显随着4-VP 相对物质的量含量的增加而减小。当nAN∶n4-VP=98∶2 时,所得共聚物的重均相对分子量达到13×104,而当4-VP相对含量增加至nAN∶n4-VP=90∶10时,所得共聚物的重均相对分子量仅为7×104。因此在单体转化率几乎相同的情况下,AN/4-VP 辐射溶液共聚合所得聚合物溶液黏度随4-VP 相对含量的增加而下降,是由于所合成的共聚物相对分子量下降所导致。这可能是因为4-VP 的竞聚率也比AN的竞聚率高[48],4-VP自由基引发AN单体的速率低于AN 自由基引发4-VP 的速率。因而当4-VP 相对含量增加时,聚合反应速率降低,在相同的辐照条件下,生成的共聚物相对分子量也较小。
辐射溶液聚合所合成的P(AN-co-4-VP)二元共聚物(图7)水接触角随4-VP 相对含量的变化如图8 所示,聚合物的水接触角随着共聚物中4-VP 相对含量的增加而逐渐减小,证明AN 与4-VP的共聚确实可以改善共聚物的亲水性。
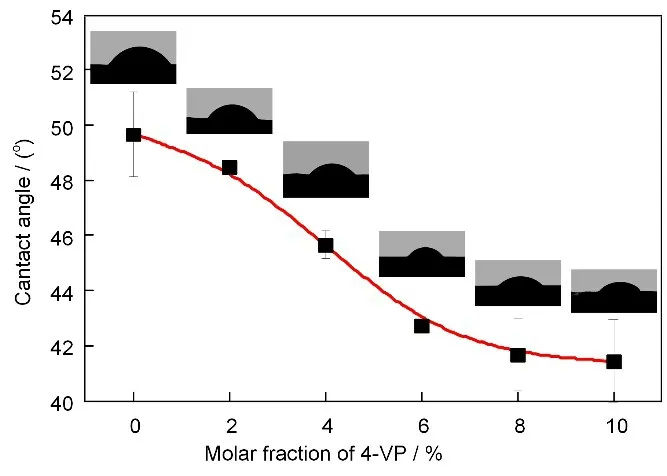
图8 AN/4-VP辐射溶液共聚合所得聚合物膜的水接触角随共聚单体相对物质的量比的变化(溶剂:DMSO;剂量率:11 Gy/min;吸收剂量:30 kGy;总单体质量分数:20%)Fig.8 Effects of the molar fraction of 4-VP on the water contact angle of P(AN-co-4-VP)prepared by the γ-ray radiation induced copolymerization of AN and 4-VP in DMSO.(Dose rate:11 Gy/min;absorbed dose:30 kGy;monomer mass fraction:20%)
3 结论
本文对γ射线辐射引发AN/4-VP在DMSO溶液中的均相共聚反应进行了研究。结果表明:γ射线辐射可以在室温下顺利引发AN 和4-VP 在DMSO中的共聚反应,吸收剂量在25 kGy 时,单体转化率即达到90%以上,但所得共聚物相对分子量与剂量率相关。在30 kGy 吸收剂量下,采用较低的剂量率(11 Gy/min)辐照时,可以直接得到黏度为250 Pa·s 的P(AN-co-4-VP)二元无规共聚物溶液,所合成的共聚物重均相对分子量为13×104左右,两种单体结构单元组成与投料比一致;当剂量率超过20 Gy/min后,所合成的共聚物相对分子量反而下降。与此同时,所得的聚合物溶液黏度随着吸收剂量的增加一直上升。当吸收剂量过高时,所生成的聚合物的辐射交联效应导致聚合物凝胶的生成。当4-VP 与AN 的相对物质的量比较低(2%)时,合成的P(AN-co-4-VP)二元无规共聚物热性能基本与PAN 均聚物一致,4-VP 并不参与-C≡N 侧基的成环或交联反应。但P(AN-co-4-VP)二元无规共聚物的水接触角随着4-VP 含量增加而逐渐减小,说明4-VP 的引入可以提高PAN的亲水性。本论文不仅为聚丙烯腈亲水改性提供了新思路,而且证明采用辐射溶液聚合方式可以直接得到高浓度(>20%)的丙烯腈均聚物或共聚物的DMSO 溶液,有望直接用于后续的成膜或纺丝工艺,这也拓展了辐射技术在丙烯腈溶液聚合领域中的应用。
作者贡献说明汪谟贞和葛学武提出了本文的研究思路和实验方案。孙锐完成了本工作中所有聚丙烯腈及其共聚物的合成、产率以及聚合物基本结构参数的表征。王雨松指导并完成聚合物的核磁共振氢谱和碳谱的测定及分析。姜志文为确定辐射聚合条件提供了指导。舒晶晶对聚合物接触角数据的测定作出了贡献。所有作者均已阅读并认可该论文最终版的所有内容。
猜你喜欢
三联生活周刊(2017年48期)2017-11-25
分析化学(2017年4期)2017-04-14
课堂内外·教师版(2017年3期)2017-04-13
现代商贸工业(2017年1期)2017-03-28
中国民族民间医药·下半月(2017年2期)2017-03-20
中学化学(2015年2期)2015-06-05
新课程·中学(2014年7期)2014-10-24
理科考试研究·高中(2014年8期)2014-10-17
农业研究与应用(2014年4期)2014-08-15
中学化学(2014年1期)2014-04-23
