
MLPA技术在α-珠蛋白生成障碍性贫血罕见基因型患者及家系分析中的应用*
2022-03-02 02:42:20刘发娣周红平柯江维
国际检验医学杂志 2022年4期
刘发娣,卢 娟,周红平,王 珂,胡 蓉,柯江维△
1.江西省儿童医院检验科,江西南昌 330006;2.南昌大学医学部基础医学院,江西南昌 330006
α-珠蛋白生成障碍性贫血(简称地贫)是人类常见的单基因遗传病之一[1],临床以小细胞低色素性贫血为主要表现,临床表型包括几乎无症状的携带者至致死性溶血性贫血患者[2]。α-地贫主要分布在热带和亚热带地区,在中国,则以广西、广东、福建、江西、四川和海南为主,地域性强[3-8]。α-地贫是由于编码珠蛋白α链的α-珠蛋白基因簇出现片段缺失或(和)点突变而引起,该基因簇位于人类染色体16p13.3,健康人每条16号染色体均有两个α-珠蛋白基因。α-地贫患者的临床表型与受累的α-珠蛋白基因个数直接相关,当一条染色体中两个α-珠蛋白基因均失活时,为α0-地贫;而一条染色体中任意失活一个α-珠蛋白基因则称为α+-地贫;两条染色体仅失活一个α-珠蛋白基因的携带者往往表现为没有临床症状的静止型;而失活两个α-珠蛋白基因的患者主要表现为轻度贫血;若4个α-珠蛋白基因中有3个基因受累则表现为血红蛋白H病;4个α-珠蛋白基因全部受累表现为Hb Bart′s胎儿水肿综合征。
目前,珠蛋白基因检测最常用的方法是跨越断裂点聚合酶链反应(Gap-PCR)和反向斑点杂交技术。在我国,引起α-地贫的突变类型包括缺失型突变和点突变,--SEA是最常见的缺失突变类型,其次为-α3.7和-α4.2;而点突变则以αQS(Hb Quong Sze)、αCS(Hb Constant Spring)和αWS(Hb Westmead)为主。本文将阐述运用多重连接探针扩增(MLPA)技术对α-珠蛋白基因罕见的27.6 kb大片段缺失突变家系的检测分析过程,而该突变为首次在江西籍人群中被发现并报道。
1 资料与方法
1.1一般资料 先证者,男,5岁9个月,江西景德镇人,因发现贫血1年半来江西省儿童医院就诊。入院时,贫血面容,皮肤无黄染,口唇淡红,口腔黏膜正常,未触及肝脾肿大。患儿父亲既往体健,系退伍军人,无贫血症状。患儿母亲贫血面容,怀孕期间曾3次晕倒。患儿有两个姐姐,均体健,无贫血表现。
1.2方法
1.2.1标本采集与血液学分析 采集患儿及其家系成员外周血2 mL(EDTA-K2抗凝),红细胞相关参数分析采用血细胞分析仪(XN-1000,日本Sysmex公司);血红蛋白电泳及组分定量分析采用快速自动电泳分析系统(Capillarys Sebia-2,法国Sebia公司)。外周血基因组DNA提取采用外周血DNA提取试剂盒(广东凯普生物技术有限公司)。
1.2.2反向斑点杂交 试验采用广东凯普生物技术有限公司的地贫检测试剂盒(含α-地贫常见缺失突变、α-地贫常见点突变和β-地贫的常见点突变),严格按照说明书操作。
1.2.3MLPA 采用P140-C1 HBA试剂(MRC-HOLLAND,荷兰),委托中翰金诺医学检验所进行检测。
1.2.4Gap-PCR 根据MLPA检测结果,参考文献[9],委托上海捷瑞生物公司合成引物(F:5′-ATCTGAGGTGGCACACAAGCA-3′;R:5′-AGCCCCAA GTCATGGACTCAC-3),运用Gap-PCR对患者及其家系成员进行扩增,引物水平为10 μmol/L,扩增体系为20 μL:其中2×PCR mix 10 μL,引物F 1 μL,引物R 1 μL,DNA模板2 μL,ddH2O 6 μL。扩增反应条件:95 ℃,3 min,1个循环;95 ℃ 50 s,62 ℃ 1 min,72 ℃ 2 min,35个循环;72 ℃ 10 min。PCR产物用2%琼脂糖凝胶电泳进行检测,同时将先证者PCR产物送广东凯普生物技术有限公司进行基因测序。
2 结 果
2.1血液学分析 先证者及其家系成员的地贫筛查血液学指标如表1所示,先证者及其母亲平均红细胞体积(MCV)和平均红细胞血红蛋白量(MCH)均显著降低,为典型的小细胞低色素性贫血,且血红蛋白A2(HbA2)均偏低,提示先证者及其母亲可能是α-珠蛋白基因突变携带者;而先证者父亲及其两个姐姐血液学指标未见明显异常。
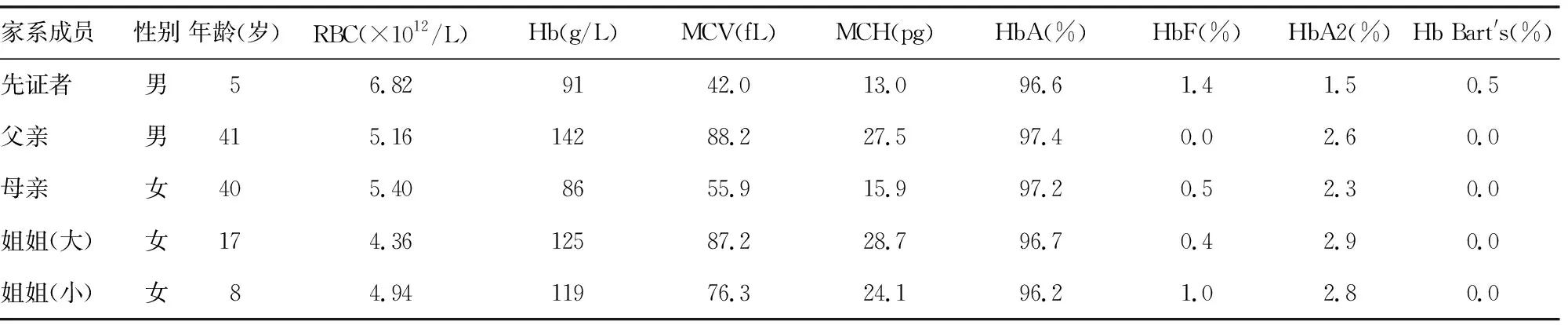
表1 先证者及其家系成员的血液学和生化参数
2.2反向斑点杂交 如图1所示,先证者α-地贫缺失突变对照点NP和点突变的正常对照点均未显示,先证者父亲未见常见的α-地贫缺失突变和点突变,先证者携带的--SEA缺失突变来源于母亲,而先证者两个姐姐的反向斑点杂交结果均未见异常。
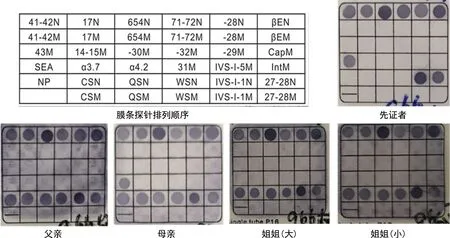
图1 反向斑点杂交法膜条探针排列顺序及先证者家系成员检测结果
2.3MLPA检测结果 先证者样本自346 nt探针至400 nt探针处均出现不同程度缺失,该缺失区域包含HBZ、HBZP1、HBAP2、HBAP1、HBA2、HBA1及HBQ1基因。根据试剂说明书可知184 nt探针至400 nt探针缺失为--SEA,包含HBAP1、HBA2、HBA1及HBQ1基因,而346 nt探针至337 nt探针缺失为-α27.6。见图2。

注:A为MLPA技术检测HBA基因的探针分布位置,灰色横条代表- -SEA缺失的范围(探针184~400 nt),黑色横条代表-α27.6缺失的范围(探针346~337 nt);B为先证者不同探针位置的拷贝数比值,其中比值在0.7~1.3为拷贝数正常,0.3~<0.7为杂合缺失,比值为0则是纯合缺失,比值>1.3代表重复。
2.4Gap-PCR和基因测序结果 根据MLPA结果,于-α27.6缺失的两侧设计引物,通过Gap-PCR扩增,将产物在2%琼脂糖凝胶中电泳,如图3所示,1、2、5这3个泳道可见约1.7 kb的特异性条带,分别是先证者及其父亲和姐姐(小);而3、4泳道显示,先证者母亲和姐姐(大)未携带该缺失突变。为了进一步明确患者的基因缺失位置,采用Sanger测序对Gap-PCR扩增产物进行测序,结果见图4,缺失的范围位于NG_000006.1:9079与NG_000006.1:36718之间,同时,两个断点之间还插入了AATCCC 6个碱基。
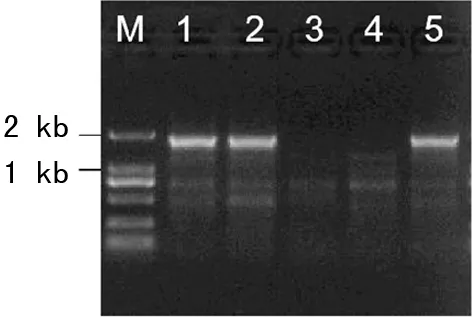
注:M为标志物;1为先证者;2为父亲;3为母亲;4为姐姐(大);5为姐姐(小);1.7 kb条带为-α27.6缺失的特异性条带。
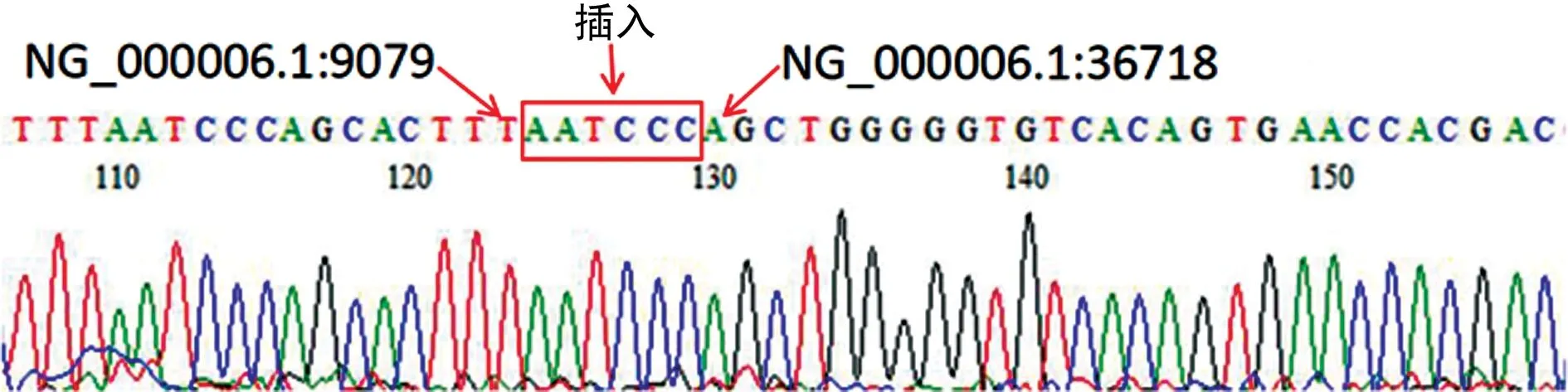
注:缺失范围见图中标识,缺失碱基T与A之间插入了6个碱基。
3 讨 论
α-地贫为最常见的常染色体隐性遗传病,随着基因检测和诊断技术的不断进步,越来越多罕见的α-地贫突变类型不断被发现。目前,检测α-地贫缺失突变最常用的方法是Gap-PCR[10],结合琼脂糖凝胶电泳或者反向斑点杂交实验共同完成。临床推广的检测试剂主要包括α-地贫的3种常见缺失类型和3种常见的点突变类型,涵盖了国内约98%的α-地贫类型[11]。本文中的先证者在常规斑点杂交实验中显示,除了携带--SEA外,缺失突变的正常对照点以及3个点突变的正常对照点均没有显色,由于本实验室所使用的检测试剂中,α-缺失突变的正常对照点的同源序列来自α2基因,而3种常见的点突变也存在于α2基因中,因此,可以推测患者出现的罕见斑点杂交图是因为患者一条染色体携带了--SEA缺失,而另一条染色体可能缺失了α2基因。
为了明确患儿所携带的α-珠蛋白基因突变型,本文选用MLPA技术进行检测,MLPA技术是2002年由SCHOUTEN等[12]首先报道,该方法结合了DNA探针杂交和PCR扩增技术,可对待检DNA序列进行定性和半定量分析,1次反应可检测45个靶序列,适用于基因片段缺失或重复以及已知点突变的检测,用于检测已知与未知的大片段缺失型α-珠蛋白基因。MLPA技术检测结果显示,先证者所携带的是一种罕见的-α27.6缺失突变。
跨过-α27.6缺失范围,在两侧设计引物,通过Gap-PCR可成功扩增出特异性条带。根据该家系的电泳结果可以看出,先证者的-α27.6缺失突变来自父亲,先证者的姐姐(小)也携带了该突变。通过对Gap-PCR产物进行Sanger测序,可准确判定缺失基因的断点,将测序结果与NCBI GeneBank比对后发现,该家系的缺失突变与此前报道的多个携带者为同一突变类型[9,13-14]。尽管目前有多篇研究报道了-α27.6缺失基因型携带者或家系,但患者均来自中国福建省[9,13-14]。本文的患者为江西省景德镇人,是江西省首次检测到该突变的携带者,丰富了江西省地贫疾病突变谱,同时也为罕见α-地贫的基因诊断方法提供参考。
不同的突变类型导致患者贫血的临床表型和机制可能不同[15],本文中先证者的父亲虽然也是-α27.6缺失基因型携带者,但他没有血液学异常的表现,并且常规临床检测试剂又未能包含该缺失类型,因此,对于地贫的罕见基因型的发现与诊断,家系分析就显得尤为重要。自从20世纪80年代开始,我国严格执行计划生育政策后,大量独生子女的出现导致大家系明显减少,也使得此类罕见型突变的发现更加困难。就本文所研究的家系而言,患儿父母虽然同时携带α-珠蛋白基因缺失突变,但是只有患儿同时遗传了父母双方的地贫缺陷基因时,才会在常规筛查实验中表现出异常的斑点杂交图,而下一代中同时携带父亲和母亲两种缺陷基因的概率仅为25%。事实也正如此,该患儿为家系中的第3个孩子,姐姐(大)珠蛋白基因未见异常,姐姐(小)则携带了与其父亲一样的罕见缺失突变,若没有先证者的出生,那么该家系所携带的27.6 kb缺失突变也难以被发现。这可能是-α27.6缺失突变携带者罕见的原因之一。因此,笔者认为-α27.6缺失突变的人群携带率可能远比实际发现的高。
综上所述,对于罕见的α-地贫突变类型,特别是尚未报道的缺失突变类型,MLPA技术是一个良好的检测手段。因此,当临床发现可疑大片段缺失的地贫患者时,可先选择MLPA技术对α-珠蛋白基因进行检测,寻找可能出现缺失的部位,再针对基因缺失部位的两端设计引物,进行Gap-PCR扩增和基因测序,便可进一步明确患者的基因型[9,16]。但是对于该家系先证者缺失了3个α基因,却没有血红蛋白H带(HbH)这一现象,虽然有文献[17]谈及,但具体的机制还不明确,还需要进一步研究探讨。
猜你喜欢
临床输血与检验(2022年3期)2022-06-22 02:52:50
检验医学与临床(2021年14期)2021-07-29 07:40:24
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
传染病信息(2019年2期)2019-05-17 13:16:04
广东海洋大学学报(2015年4期)2016-01-13 08:39:30
听力学及言语疾病杂志(2015年5期)2015-12-24 01:47:04
首都医科大学学报(2015年4期)2015-12-16 13:00:08
检验医学与临床(2015年14期)2015-03-16 01:46:53
重庆医学(2015年12期)2015-03-05 05:52:54
郑州大学学报(医学版)(2015年2期)2015-02-27 14:50:44
