
深圳地区两种类型的珠蛋白生成障碍性贫血的基因型及表型研究
2015-03-16 01:46:53裴元元李高驰魏凤香
检验医学与临床 2015年14期
裴元元,李高驰,冉 健,魏凤香
(广东省深圳市龙岗区妇幼保健院 518172)
·论 著·
深圳地区两种类型的珠蛋白生成障碍性贫血的基因型及表型研究
裴元元,李高驰,冉 健,魏凤香△
(广东省深圳市龙岗区妇幼保健院 518172)
目的 分析深圳地区人群的αβ复合型珠蛋白生成障碍性贫血及非缺失型α-珠蛋白生成障碍性贫血的血液学特征及基因突变类型。方法 收集2013年5月至2014年5月血液学及血红蛋白电泳筛查阳性的3 082例疑似珠蛋白生成障碍性贫血患者,检测中国人群中最常见的 17 种β-珠蛋白生成障碍性贫血突变、3种α-珠蛋白生成障碍性贫血点突变及3种缺失型α-珠蛋白生成障碍性贫血基因改变。结果 1 042例经基因确诊的珠蛋白生成障碍性贫血患者中有αβ复合型珠蛋白生成障碍性贫血35例,非缺失型α-珠蛋白生成障碍性贫血60例。αβ复合型珠蛋白生成障碍性贫血表现为小细胞低色素及血红蛋白(Hb)A2的升高,β-珠蛋白生成障碍性贫血基因以CD41-42 (37.1%)、IVS-2-654 (31.4%)、-28 (14.3%)突变较为常见,α-珠蛋白生成障碍性贫血基因则以--SEA(42.1%)为主;非缺失型α-珠蛋白生成障碍性贫血中,αCSα/αα,αQSα/αα表现为小细胞低色素,αWSα/αα携带者则无血液学异常表现,αCSα/αα伴有Hb A2的降低,Hb CS、Hb WS、Hb QS的构成比依次为54.1%、31.1%、14.8%。两类患者的红细胞、平均红细胞Hb浓度、红细胞体积分布宽度均在正常值范围。结论 深圳地区αβ复合型珠蛋白生成障碍性贫血发生率为1.14%,非缺失型α-珠蛋白生成障碍性贫血发生率为1.9%,两类患者均缺乏特异性的血液学指标变化。
αβ复合型珠蛋白生成障碍性贫血; 非缺失型α-珠蛋白生成障碍性贫血; 基因型; 血液学表型
珠蛋白生成障碍性贫血在临床上根据合成障碍的肽链不同,大致分为α-珠蛋白生成障碍性贫血和β-珠蛋白生成障碍性贫血两大类;根据α-和β-链数量不平衡程度分为轻型、中间型、重型3种,轻型和中间型珠蛋白生成障碍性贫血患者一般可生存到成年期,重型患者常胎死宫内或在幼年期夭折。该病在我国广西、广东的发生率最高。据报道,广州地区α-珠蛋白生成障碍性贫血的发生率约为8.3%,β-珠蛋白生成障碍性贫血的发生率约为2.4%[1-2]。这两类杂合子婚配即有可能出生αβ复合型珠蛋白生成障碍性贫血后代,αβ复合型珠蛋白生成障碍性贫血患者无论是与α-珠蛋白生成障碍性贫血或β-珠蛋白生成障碍性贫血携带者结合,都会有1/4的概率生育重型珠蛋白生成障碍性贫血患儿[3]。另外,由于非缺失型α-珠蛋白生成障碍性贫血的绝大多数突变位于功能较强的α2基因,α2基因发生点突变时,α肽链的产量比α2基因发生缺失时明显减少,非缺失型α-珠蛋白生成障碍性贫血引起的血红蛋白(Hb)H病临床表现更为严重。因此αβ复合型珠蛋白生成障碍性贫血及非缺失型α-珠蛋白生成障碍性贫血的准确检出对于做好优生优育工作尤为重要。本文就临床实践中发现的αβ复合型珠蛋白生成障碍性贫血和非缺失型α-珠蛋白生成障碍性贫血的血液表型以及基因型进行综合分析,旨在掌握本地区人群中αβ复合型珠蛋白生成障碍性贫血和非缺失型α-珠蛋白生成障碍性贫血的基因型分布、血液表型等资料,为制订可行有效的珠蛋白生成障碍性贫血干预方案提供科学依据。
1 资料与方法
1.1 一般资料 选择2013年5月至2014年5月本院收治的3 082例疑似珠蛋白生成障碍性贫血患者。
1.2 方法
1.2.1 珠蛋白生成障碍性贫血筛查 取静脉血2 mL,乙二胺四乙酸二钾(EDTA-K2)抗凝,东亚Sysmex血细胞分析仪分析红细胞参数,如红细胞计数(RBC)、Hb、平均红细胞体积(MCV)、平均红细胞血红蛋白量(MCH)、平均红细胞血红蛋白浓度(MCHC)、红细胞体积分布宽度(RBC-SD)。MCV≤80 fL和(或)MCH≤27 pg者使用Sebia全自动毛细管电泳仪进行Hb电泳,分析Hb成分及其百分比,Hb A2<2.5% 怀疑α-珠蛋白生成障碍性贫血,Hb A2>3.5%则怀疑β-珠蛋白生成障碍性贫血,对疑似珠蛋白生成障碍性贫血患者进一步进行珠蛋白生成障碍性贫血基因诊断。
1.2.2 珠蛋白生成障碍性贫血基因检测 采用深圳益生堂全血基因提取试剂盒提取外周血基因组DNA,基因诊断试剂盒诊断α-珠蛋白生成障碍性贫血的3种缺失型突变(gap-PCR技术)和3种点突变(PCR结合寡核苷酸探针反向印迹杂交法)以及β-珠蛋白生成障碍性贫血的17种点突变(PCR/寡核苷酸探针反向印迹杂交法)。
2 结 果
2.1 珠蛋白生成障碍性贫血基因检测结果 3 082例疑似患者共有1 042例确诊为珠蛋白生成障碍性贫血。单纯性α-珠蛋白生成障碍性贫血622例(59.7%),单纯性β-珠蛋白生成障碍性贫血385例(36.9%),αβ复合型珠蛋白生成障碍性贫血35例(3.4%)。
2.2 αβ复合型珠蛋白生成障碍性贫血
2.2.1 红细胞参数分析 以MCV<80 fL,MCH<27 pg为标准,此类患者主要表现为小细胞、低色素。以轻度贫血Hb 90 g/L至正常下限,中度贫血Hb 60~89 g/L,重度贫血Hb 30~59 g/L,极重度贫血Hb<30 g/L为标准,此类患者主要表现为轻度贫血,其余红细胞参数RBC、MCHC、RBC-SD均在正常值范围,见表1。
2.2.2 Hb电泳分析 35例复合型珠蛋白生成障碍性贫血患者未发现有Hb F异常,Hb A2水平均在5%以上。见表1。
2.2.3 αβ复合型珠蛋白生成障碍性贫血的基因型及其频率 共发现32例αβ双杂合突变,2例Hb H合并β珠蛋白生成障碍性贫血杂合突变--SEA/-α3.7合并IVS-2-654、--SEA/αWSα合并IVS-2-654以及1例αCSα/-α3.7合并CD17。35例复合型珠蛋白生成障碍性贫血患者中,α-珠蛋白生成障碍性贫血基因常见的3种缺失型突变和3种点突变均有检出,其中--SEA(42.1%)最常见,其次是-α3.7(26.3%)和-α4.2(13.2%)。共检出7种β-珠蛋白生成障碍性贫血基因突变类型,分别为CD41-42(37.1%)、IVS-2-654(31.4%)、-28(14.3%)、CD17(8.6%)、-29(2.9%)、CD71-72(2.9%)、IVS-1-1(2.9%)。构成18种β-珠蛋白生成障碍性贫血杂合突变并发α-珠蛋白生成障碍性贫血的复合基因型。见表2。
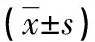
表1 珠蛋白生成障碍性贫血各突变类型的红细胞参数分析
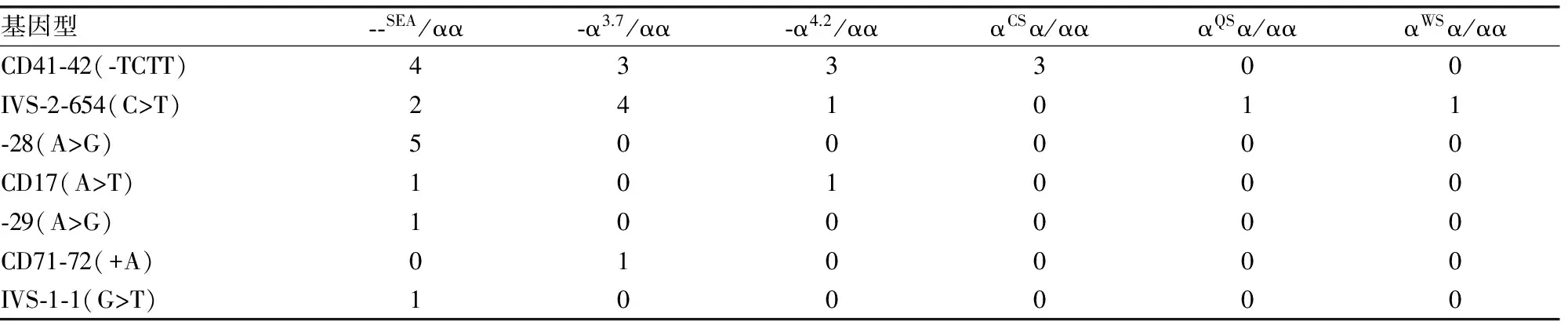
表2 32例αβ复合型珠蛋白生成障碍性贫血的基因型分布(n)
2.3 非缺失型α-珠蛋白生成障碍性贫血
2.3.1 红细胞参数分析 αCSα/αα,αQSα/αα表现为小细胞低色素,除αQSα/αα患者Hb轻度降低外,其余红细胞参数RBC、MCHC、RBC-SD均在正常值范围。αWSα/αα携带者则无血液学异常表现。见表1。
2.3.2 Hb电泳分析 发现3例αCSα/αα及1例αWSα/αα患者的Hb F大于2.5%,其余非缺失型α-珠蛋白生成障碍性贫血患者未见Hb F异常。所有αQSα/αα及αWSα/αα患者的Hb A2均在正常范围(2.5%~3.5%),而αCSα/αα患者的Hb A2均小于2.5%,且有14例αCSα/αα患者检出有Hb CS条带,含量在0.4%~1.3%。
2.3.3 非缺失型α-珠蛋白生成障碍性贫血基因型及其频率 622例单纯性α-珠蛋白生成障碍性贫血患者中,单纯性非缺失型α-珠蛋白生成障碍性贫血47例(αCSα/αα 27例,αWSα/αα 14例,αQSα/αα 6例),αQSα/αWSα 1例,αWSα/--SEA2例,αCSα/--SEA1例,αQSα/--SEA1例,αCSα/-α4.21例。累加3例αCSα/αα合并CD41-42、1例αQSα/αα合并IVS-2-654、1例αWSα/αα合并IVS-2-654、1例--SEA/αWSα合并IVS-2-654及1例αCSα/-α3.7合并CD17,推算本地区非缺失型α-珠蛋白生成障碍性贫血携带率为1.9% (60/3 082),Hb CS、Hb WS、Hb QS构成比依次为54.1%、31.1%、14.8%。
3 讨 论
α-珠蛋白生成障碍性贫血主要是由于α珠蛋白基因的大片段丢失致使α珠蛋白量下降,体内过多的γ珠蛋白或β珠蛋白形成Hb Bart′s或Hb H,导致无效造血和红细胞被破坏;也有少量α-珠蛋白生成障碍性贫血是由于α1或α2基因的点突变引起。β-珠蛋白生成障碍性贫血主要是由于β基因点突变使β珠蛋白合成减少,多余的α珠蛋白沉积在红细胞膜上,造成红细胞破坏。目前,大部分遗传病尚无有效的治疗方法,大规模筛查突变基因携带者,阻止患儿出生是必要的预防措施。
流行病学调查显示,广东省人群中同时携带α和β-珠蛋白生成障碍性贫血基因突变的比例为 0.26%[4],中山市人群αβ复合型珠蛋白生成障碍性贫血发生率为1.58%[5]。本研究中,深圳市人群αβ复合型珠蛋白生成障碍性贫血的发生率为1.14%(35/3 082),高于全省平均水平[4,6]。35例复合型珠蛋白生成障碍性贫血患者中共检出18种β-珠蛋白生成障碍性贫血基因杂合突变合并α-珠蛋白生成障碍性贫血基因突变的复合基因型,其中β-珠蛋白生成障碍性贫血基因以CD41-42 (37.1%)、IVS-2-654 (31.4%)、-28 (14.3%)杂合突变较为常见,α-珠蛋白生成障碍性贫血基因则以--SEA(42.1%)为主,β基因CD 41-42突变与α基因--SEA/αα突变恰是深圳地区最常见突变类型[7]。在血液学上,该类患者有小细胞低色素表现,但其贫血并不严重,多为轻度贫血或正常,RBC、MCHC、RBC-SD等其他红细胞参数均在正常值范围。Hb电泳结果仅表现为Hb A2的升高,与单纯的β-珠蛋白生成障碍性贫血携带者较为相似。本研究发现的2例β-珠蛋白生成障碍性贫血复合Hb H病(--SEA/-α3.7合并IVS-2-654、--SEA/αWSα合并IVS-2-654)以及1例αCSα/-α3.7合并CD17患者的Hb电泳也均未发现Hb H 或Hb Bart′s,这可能是由于α和β-珠蛋白基因同时有缺陷,导致α链和β链的合成同时减少,从而使α链与β链的相对不平衡减轻,不出现多余β链和γ链的聚合。由于复合型珠蛋白生成障碍性贫血血液学改变没有其独特的性质,因此仅使用MCV、MCH等血液学指标及Hb电泳筛查复合型珠蛋白生成障碍性贫血可能出现漏诊或误诊,在临床工作中一定要加强对αβ复合型珠蛋白生成障碍性贫血的重视,对β-珠蛋白生成障碍性贫血筛查阳性的患者,建议其同时进行α和β-珠蛋白生成障碍性贫血基因诊断,以便正确地指导遗传咨询和产前诊断。
少部分由于α珠蛋白基因点突变而引起的α-珠蛋白生成障碍性贫血即称为非缺失型α-珠蛋白生成障碍性贫血。正常人每条16号染色体上有α1和α2两个高度同源的α珠蛋白基因,α2较α1基因功能强大,其表达量约是α1基因的两倍,α2基因的突变对降低基因产物的作用更大。目前,全世界已经报道了70多种不同的非缺失型α基因突变,大多数的突变位于α2基因内[8]。有资料显示,不同地区常见的非缺失型α-珠蛋白生成障碍性贫血基因突变的携带率及构成比不同[9]。本研究中发现,深圳市非缺失型α-珠蛋白生成障碍性贫血携带率为1.9%,Hb CS、Hb WS、Hb QS的构成比依次为54.1%、31.1%、14.8%,符合我国南方非缺失型α-珠蛋白生成障碍性贫血的人群分布特点[10]。在血液学上,αCSα/αα,αQSα/αα表现为小细胞低色素,轻度贫血或正常,红细胞参数RBC、MCHC、RBC-SD均在正常值范围。αWSα/αα携带者则无血液学异常。Hb电泳结果显示所有αQSα/αα及αWSα/αα患者的Hb A2含量均在正常范围,而αCSα/αα患者的Hb A2均小于2.5%,且仅有部分患者可以检测出Hb CS条带。血液学指标对非缺失型α-珠蛋白生成障碍性贫血携带者的正确检出作用不大。非缺失型α-珠蛋白生成障碍性贫血虽然不是珠蛋白生成障碍性贫血的主要突变类型,但其所占的比例不容忽视,在α-珠蛋白生成障碍性贫血高发区准确筛查出非缺失型α-珠蛋白生成障碍性贫血,对预防具有更严重临床表现的非缺失型α-珠蛋白生成障碍性贫血胎儿(αCSα/--SEA、αQS/--SEA、αWSα/--SEA等)的出生极其重要。
目前珠蛋白生成障碍性贫血尚无确切有效的治疗方法,在临床工作中一定要加强对αβ复合型珠蛋白生成障碍性贫血及非缺失型α-珠蛋白生成障碍性贫血的重视,减少中间型、重型珠蛋白生成障碍性贫血患儿的发生率,提高人口素质。
[1]杜传书.地中海贫血研究的现状与未来[J].中华医学遗传学杂志,1996,13(5):257-258.
[2]Xu XM,Zhou YQ,Luo GX,et al.The prevalence and spectrum of alpha and beta thalassaemia in Guangdong Province:implications for the future health burden and population screening[J].J Clin Pathol,2004,57(5):517-522.
[3]蔡永林,郑裕明,汤敏中,等.β-地中海贫血复合缺失型α-地中海贫血双重杂合子的分子检测及血液学分析[J].中国实验血液学杂志,2007,15(1):195-197.
[4]李莉艳,李强,宋兰林,等.69例αβ复合型地中海贫血的血液学和基因型研究[J].实用妇产科杂志,2011,27(5):378-381.
[5]黄道连,袁春雷,冯丹艺.αβ复合型地中海贫血筛查结果分析[J].中国小儿血液与肿瘤杂志,2011,16(5):214-216.
[6]韩俊英,曾瑞萍,胡彬.广东地区β地中海贫血复合α缺失型地中海贫血双重杂合子检出率[J].中华血液学杂志,2001,22(12):514-516.
[7]袁晖,吴维青,吴晓霞,等.深圳地区育龄人群地中海贫血基因型分布调查[J].中山大学学报:医学科学版,2012,33(4):553-557.
[8]Harteveld CL,Higgs DR.Alpha-thalassaemia[J].Orphanet J Rare Dis,2010,5(10):13.
[9]潘干华,申芫子,黄勇,等.广东南海地区非缺失型α地中海贫血分子流行病学调查[J].国际检验医学杂志,2014,1(35):56-57.
[10]段山,李洪义,陈争,等.中国南方α-地中海贫血基因突变型研究[J].中国实验血液学杂志,2003,11(1):54-60.
(收稿日期:2015-02-25 修回日期:2015-03-18)
Study on genotypes and phenotypes of αβ compound thalassemia and non-deletion α thalassemia in Shenzhen area
*PEIYuan-yuan,LIGao-chi,RANJian,WEIFeng-xiang△
(LonggangDistrictMaternalandChildHealthCareHospital,Shenzhen,Guangdong518172,China)
Objective To analyze the hematological features and types of gene mutation of αβ compound thalassemia and non-deletion α thalassemia in Shenzhen area.Methods 3 082 patients with suspected thalassemia screened by the hematological and hemoglobin electrophoresis screening in our hospital from May 2013 to May 2014 were collected and detected the gene changes in the commonest 17 kinds of β thalassemia mutation,3 kinds of α thalassemia point mutation and 3 kinds of deletion α thalassemia among Chinese population.Results Among 1 042 cases of thalassemia definitely diagnosed by gene,35 cases were αβ compound thalassemia and 60 cases were non-deletionα thalassemia.αβ compound thalassemia was manifested by microcytic hypochromic anemia and the increase of Hb A2.The mutations of CD41-42(37.1%),IVS-2-654 (31.4%) and-28 (14.3%) in β thalassemia gene were common,while the α thalassemia gene was dominated by--SEA(42.1%);in non-deletion αthalassemia,αCSα/αα and αQSα/αα were manifested by microcytic and hypochromic,αWSα/αα carriers had no hematological abnormal manifestation and αCSα/αα was accompanied by the Hb A2 decrease,the constituent ratio of Hb CS,Hb WS and Hb QS were 54.1%,31.1% and 14.8% respectively.Conclusion The occurrence rates of αβ compound thalassemia and non-deletion α thalassemia in Shenzhen area are 1.14% and 1.9% respectively.The patients of these two kinds of thalassemia all lack the changes of specific hematological indexes.
αβ compound thalassemia; non-deletion α thalassemia; genotype; hematological phenotype
2015-02-26
2015-04-20)
国家自然科学基金项目(81201568);深圳市龙岗区科技计划项目(YS2013167)。
裴元元,女,博士研究生,主管技师,主要从事分子遗传、产前筛查诊断工作。△
,E-mail:haowei727499@163.com。
10.3969/j.issn.1672-9455.2015.14.002
A
1672-9455(2015)14-1981-03
猜你喜欢
中国毕业后医学教育(2022年1期)2022-08-19 02:51:34
检验医学与临床(2021年14期)2021-07-29 07:40:24
现代畜牧科技(2021年4期)2021-07-21 06:13:16
猪业科学(2018年8期)2018-09-28 01:28:04
检验医学与临床(2015年22期)2015-03-16 07:05:47
现代检验医学杂志(2015年1期)2015-02-06 01:59:18
中国中医药现代远程教育(2014年14期)2014-03-01 04:27:20
实验动物与比较医学(2014年3期)2014-02-28 14:52:55
河南医学研究(2014年2期)2014-02-27 14:51:33
卫生职业教育(2014年8期)2014-02-16 08:00:16
