
刺五加标准汤剂制备及质量标准研究
2022-02-16 01:06:54张金秋曹玉春魏晓雨
亚太传统医药 2022年1期
张金秋,边 雨,曹玉春,魏晓雨,邹 慧
(吉林省现代中药工程研究中心有限公司,吉林 长春 130012)
刺五加为五加科植物刺五加Acanthopanaxseuticosus(Rupr.et Maxim.)Harms的干燥根和根茎或茎。春秋二季采收,洗净,干燥。刺五加主要分布于东北三省、山西、河北等地,其中黑龙江和吉林产地为道地产区[1]。主要化学成分有黄酮、皂苷、氨基酸及有机酸等,具有益气健脾、补肾安神等功效[2-3]。相比传统汤剂的服用、储存以及不方便携带等问题,中药配方颗粒具有质量可控性、疗效可靠、服用便捷等优点[4-6]。
依据国家药典委员会发布的《中药配方颗粒质量控制与标准制定技术要求》(征求意见稿)[7],以下简称《征求意见稿》,中药配方颗粒的制备,除制剂工艺外,其余应与传统汤剂基本一致,即以水提取,以物理方法固液分离、浓缩、干燥、颗粒成型等工艺生产。中药配方颗粒药效物质应与中药饮片水煎汤剂保持基本一致[5]。
本研究收集全国主要刺五加产地样品,按照《征求意见稿》进行标准汤剂的制备,测定紫丁香苷含量,建立指纹图谱,进行系统聚类分析和主成分分析,以评价不同产地刺五加标准汤剂的质量,为后续生产配方颗粒的质量控制提供参考依据。
1 仪器与试药
1.1 仪器
高效液相色谱仪(Thermo公司,美国,型号:Ultimate3000);多锅煎药机(山东青州精诚医药设备制造有限公司,型号:JY-8);冷冻干燥机(宁波新芝生物科技股份有限公司,型号:Scientz-30);超声波清洗器(宁波新芝生物科技股份有限公司,型号:SB-1200D);薄层成像系统(上海科哲生化科技有限公司,型号:GoodLook-1000);电子天平(梅特勒-托利多仪器(上海)有限公司,型号:ME204E、AB135-S)。
1.2 试剂与试药
试剂:甲醇、乙腈为色谱纯,来自Fisher公司,其他试剂均为分析纯,水为超纯水。
对照品:原儿茶酸(批号110809-201205,纯度:99.90%)、紫丁香苷(批号111574-201605,纯度:98.8%)、绿原酸(批号110753-201716,纯度:99.3%)均购买于中国食品药品检定研究院。
1.3 样品
刺五加药材:包含了刺五加药材的道地产区及主产区,依次编号为A1-A16,具体信息见表1。

表1 16样品信息
2 方法与结果
2.1 标准汤剂的制备
参照《征求意见稿》中技术要求进行制备,方法如下:取刺五加药材饮片100 g,分别加9倍、7倍量水进行煎煮,第一次浸泡30 min,武火煮沸、文火保持微沸,煎煮时间分别为60 min、40 min,过滤,合并滤液,温度低于50 ℃减压浓缩至100 mL。将浓缩液置于培养皿中,-56 ℃冻存12 h,冷冻干燥,即得刺五加标准汤剂冻干粉。依次编号为S1-S16。如表1所示。
2.2 紫丁香苷含量测定
参照2015版《中国药典》[1]刺五加药材项下紫丁香苷含量测定方法进行测定。
2.2.1 色谱条件 以十八烷基硅烷键合硅胶为填充剂;以甲醇-水(20∶80)为流动相;检测波长为265 nm。色谱见图1。
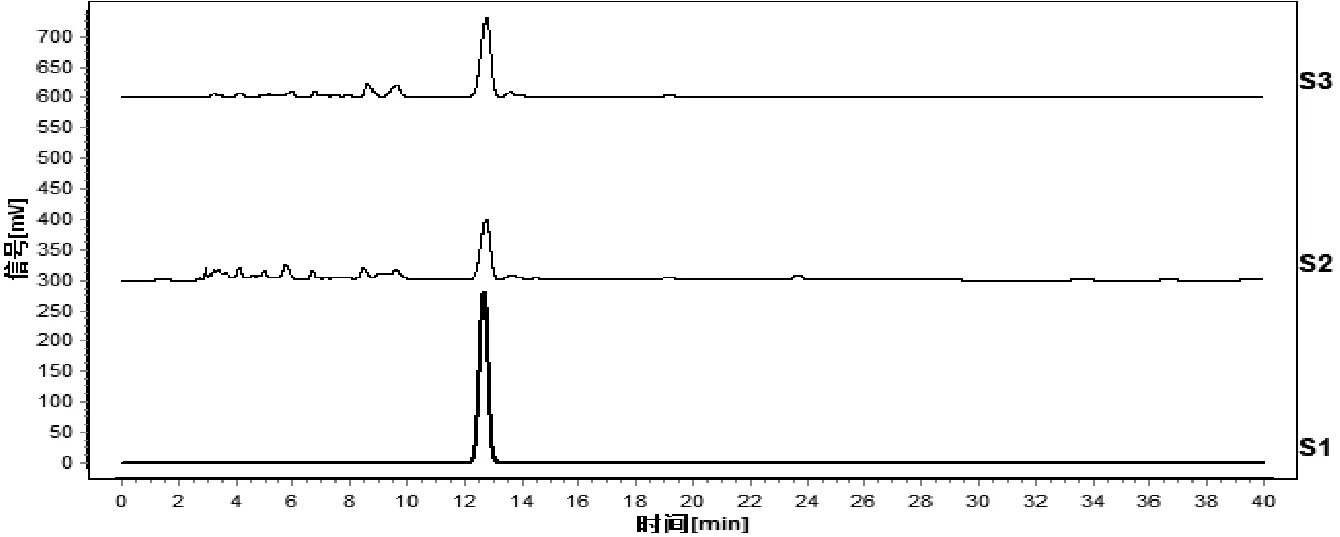
图1 对照品(S1)、药材供试品(S2)、标准汤剂供试品(S3)HPLC色谱
2.2.2 对照品溶液的制备 取紫丁香苷对照品适量,精密称定,加甲醇制成每1 mL含紫丁香苷0.50 mg的溶液。
2.2.3 供试品溶液的制备 取本品粉末约0.2 g,精密称定,置于25 mL量瓶中,加20 mL甲醇溶解,超声(功率250W,频率50 kHz)20 min,取出,冷却至室温,加甲醇至刻度,即得。
2.2.4 测定 分别精密吸取对照品溶液与供试品溶液各10 μL,注入液相色谱仪,测定,即得。
2.2.5 方法学考察 (1)线性关系的考察:精密称取紫丁香苷对照品21.94 mg(中国食品药品检定研究院,批号111574-201504,98.7%),置10 mL量瓶中,加甲醇适量,超声溶解,取出,放冷,加甲醇至刻度,摇匀,即得2.165 5 mg·mL-1的紫丁香苷对照品贮备液;分别精密移取贮备液0.125、0.25、0.5、1、2.5 mL,至5 mL量瓶中,加甲醇至刻度,得浓度分别为0.054 2、0.108 5、0.216 9、0.433 9、1.084 7 mg·mL-1的对照品溶液。按照“2.2.4”项下方法测定。以浓度(mg·mL-1)为横坐标,峰面积为纵坐标,绘制标准曲线。得回归曲线y=44.659x+0.115,相关系数r=0.999 8。结果表明,紫丁香苷在0.054 2~1.084 7 mg·mL-1范围内线性良好。
(2)精密度试验:取同一批样品(编号S1)约0.2 g,精密称定,按“2.2.3”项下方法制备供试品溶液,精密吸取供试品溶液10 μL。按“2.2.1”项下色谱条件测定,连续进样6次,结果紫丁香苷峰面积RSD=0.23%,表明仪器精密度良好。
(3)重复性试验:取同一批(编号S1)刺五加标准汤剂干粉约0.2 g,精密称定,按“2.2.3”项下方法制备6份供试品溶液,精密吸取供试品溶液10 μL。按“2.2.1”项下色谱条件测定,测得同一批标准汤剂干粉中紫丁香苷的含量均值为1.48%,RSD=1.49%(n=6),表明方法重复性良好。
(4)稳定性试验:取同一批(编号S1)刺五加标准汤剂干粉约0.2 g,精密称定,按“2.2.3”项下方法制备供试品溶液,精密吸取供试品溶液10 μL。按“2.2.1”项下色谱条件测定,分别在制备后放置0、4、8、12、16、24 h进样检测,结果紫丁香苷峰面积RSD=1.70%(n=6),表明供试品溶液在24 h内稳定。
(5)回收率试验:取已知含量的紫丁香苷标准汤剂干粉(编号S1),约0.1 g(共6份),精密称定,分别置于25 mL容量瓶中,分别精密加入对照品溶液各1 mL(对照品溶液浓度为1.462 7 mg·mL-1),加入一定量的紫丁香苷对照品,按“2.2.3”项下方法制备供试品溶液,按照“2.2.1”色谱条件进行液相分析。结果,平均回收率为98.71%,RSD为1.47%(n=6),表明本方法准确度良好。
2.2.5 指标成分含量测定 按照“2.2.1”项下色谱条件测定16批标准汤剂中紫丁香苷的含量,并计算紫丁香苷转移率,结果见表2。

表2 16批标准汤剂中紫丁香苷测定结果 (%)
结果,16批标准汤剂平均出膏率为5.74%,按照均值±30%的范围浮动为4.02%~7.46%;标准汤剂中紫丁香苷平均含量为4.41%,按照均值±30%的范围浮动为3.09%~5.73%;紫丁香苷平均转移率为42.25%,按照均值±30%的范围浮动为29.58%~54.93%,本研究的16批标准汤剂出膏率、紫丁香苷含量及转移率均在此范围内,表明制备工艺规范,稳定、可行。
2.3 指纹图谱研究
2.3.1 色谱条件 色谱柱为WondaSil C18(4.6 mm×250 mm,5 μm),以乙腈(A)-0.2%磷酸水溶液(B)为流动相;梯度洗脱,0~30 min,8%~16%乙腈;30~40 min,16%~25%乙腈。检测波长为220 nm,柱温为25 ℃,流速为0.8 mL·min-1。此色谱条件下,各色谱峰的分离度良好,峰形对称。色谱见图2。
2.3.2 混合对照品溶液的制备 取原儿茶酸、紫丁香苷、绿原酸对照品适量,加入甲醇,即得每1 mL含原儿茶酸39.6 μg、紫丁香苷80 μg、绿原酸170 μg的混合对照品溶液。
2.3.3 供试品溶液的制备 取本品粉末约0.2 g,精密称定,置于25 mL量瓶中,加20 mL甲醇溶解,超声(功率250 W,频率50 Hz)20 min,取出,冷却至室温,加甲醇至刻度,即得。
2.3.4 方法学考察 (1)精密度试验:取同一批刺五加标准汤剂干粉约0.2 g,精密称定,按照“2.3.3”方法制备供试品溶液一份,按“2.3.1”项下方法连续进样6次,以紫丁香苷(S)为参照峰,计算7个共有峰的相对峰面积,结果RSD均在3%以内,表明仪器精密度良好。
(2)重复性试验:取同一批刺五加标准汤剂干粉约0.2 g,精密称定6份,按照“2.3.3”方法制备供试品溶液,按“2.3.1”项下方法进行测定,以紫丁香苷(S)为参照峰,计算7个共有峰的相对峰面积,结果RSD均在3%以内,表明方法重复性良好。
(3)稳定性试验:取同一批刺五加标准汤剂干粉约0.2 g,精密称定,按照“2.3.3”方法制备供试品溶液,分别在制备后放置0、4、8、12、16、24 h后,按照“2.3.1”项下方法进行测定,以紫丁香苷(S)为参照峰,计算7个共有峰的相对峰面积,结果RSD均在3%以内,表明供试品溶液在24 h稳定性良好。
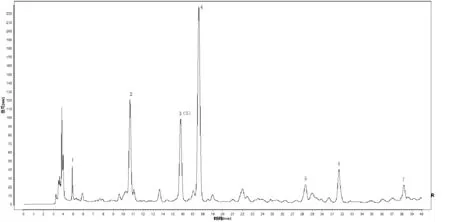
峰2:原儿茶酸;峰3(S):紫丁香苷,峰4:绿原酸。图2 刺五加标准汤剂HPLC对照指纹图谱
(4)指纹图谱的建立与分析:按照“2.3.1”项下色谱条件测定16批刺五加标准汤剂指纹图谱,将所得到的色谱数据导入《中药指纹图谱相似度评价系统(2012版)软件》,采用多点校正将谱峰自动匹配,中位数法计算得出样品指纹图谱的共有模式。刺五加标准汤剂指纹图谱中呈现7个共有峰,通过与对照品图谱比较,指认峰2为原儿茶酸,峰3为紫丁香苷,峰4为绿原酸,其中以紫丁香苷为参照峰(S峰),16批次刺五加标准汤剂HPLC指纹图谱相似度均在0.9以上。色谱见图3。
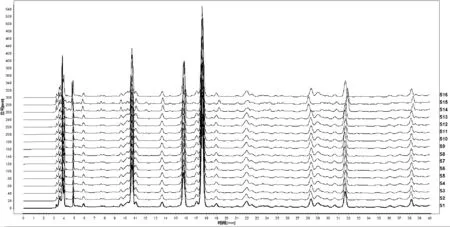
图3 16批刺五加标准汤剂HPLC色谱
2.3.5 聚类分析 将16批刺五加标准汤剂干粉(编号S1-S16)指纹图谱中的7个共有峰峰面积为变量,采用SPSS 21.0 软件,以组间平均数连接法、平方Euclideam距离为度量标准进行聚类分析。结果,16批刺五加标准汤剂干粉可聚为4类,其中S1聚为一类,S7聚为一类,S5、S8、S15聚为一类、剩余其他聚为一类,详见图4。
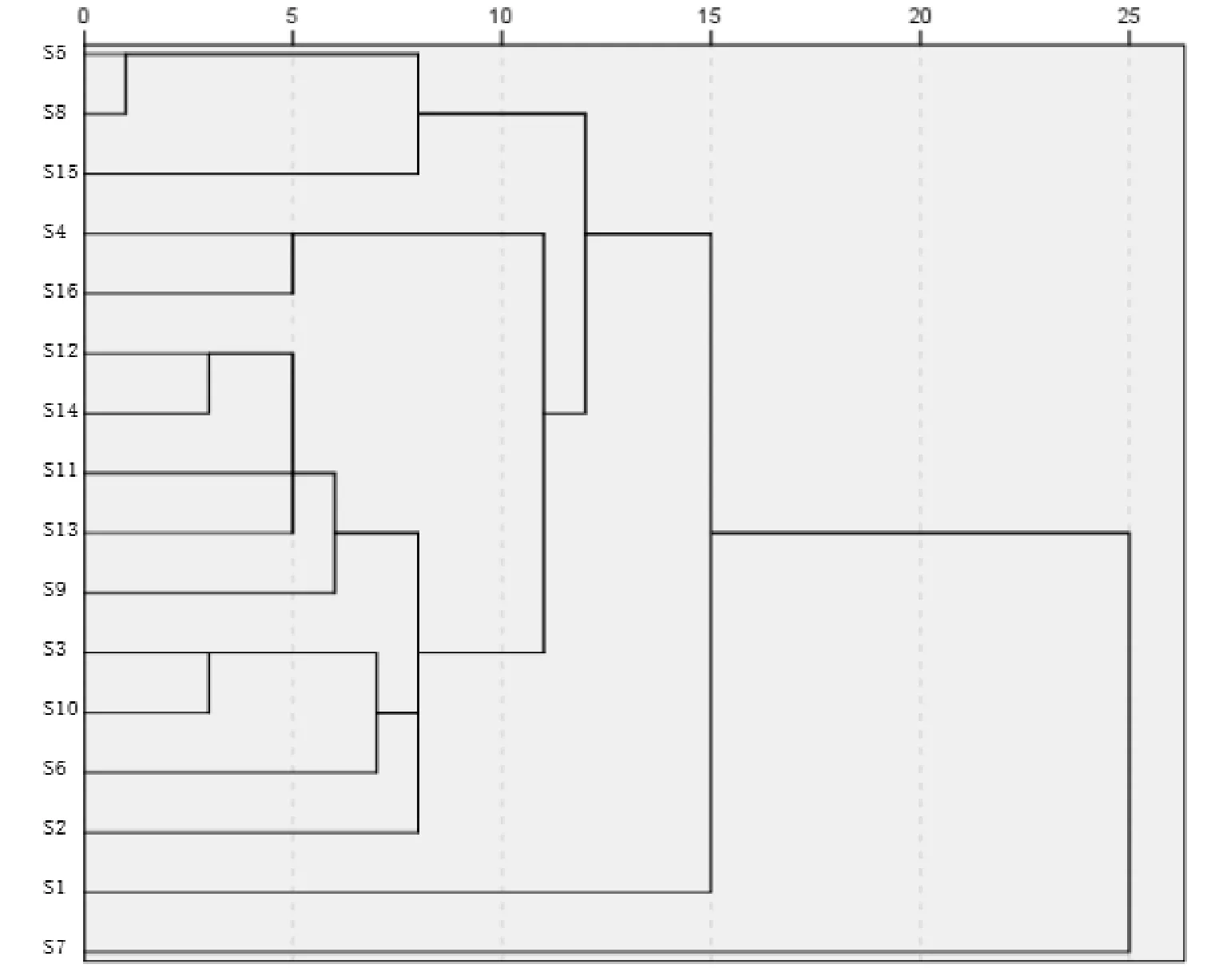
图4 16批刺五加标准汤剂干粉的聚类分析树状图
2.4 主成分分析
采用SPSS 21.0软件对16批刺五加标准汤剂干粉的7个共有峰峰面积进行主成分分析,以特征值>1为判断标准。特征值和方差贡献率见表7,由表7可知,共提取了3个主成分,3个主成分的累积方差贡献率为67.649%,详见表3。
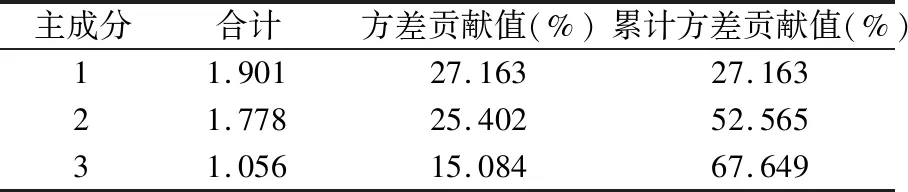
表3 特征值和方差贡献率
主成分载荷矩阵(也称主成分系数)不等同于因子载荷矩阵,可通过因子载荷矩阵计算得到,根据表3结果按公式计算,Ui=Ai/√λ[6](式中,Ui为载荷矩阵,Ai为因子载荷矩阵,λ为特征值),峰1、峰2(原儿茶酸)、峰7在第1主成分有较高载荷,峰3(紫丁香苷)、峰4(绿原酸)、峰5在第2主成分有较高载荷,峰2(原儿茶酸)在第2主成分有较高载荷,结果见表4。

表4 主成分矩阵
每一个载荷量表示主成分与对应变量的系数,第1主成分得分为Z1=0.295X1+0.466X2-0.045X3-0.060X4-0.126X5-0.599X6+0.561X7,第2主成分得分Z2=-0.005X1-0.070X2+0.601X3+0.517X4+0.571X5-0.162X6+0.120X7,第3主成分得分Z3=-0.742X1+0.378X2+0.244X3-0.367X4+0.108X5+0.174X6+0.265X7,综合得分Z=(27.163%×Z1+25.402%×Z2+15.084%×Z3)/67.649%;综合得分越高,表示质量越好。结果,16批样品中,吉林安图(S1)最优,吉林通化(S8)和黑龙江哈尔滨(S9)较佳,详见表5。

表5 16批刺五加标准汤剂综合得分结果
3 讨论
刺五加药材饮片中紫丁香苷平均含量为0.7%,平均出膏率为5.74%,标准汤剂中紫丁香苷平均含量为4.41%,从刺五加药材饮片到标准汤剂冻干粉中紫丁香苷转移率为42.25%。
中药标准汤剂是在中医药理论指导下依据临床汤剂煎煮方式规范化进行煎煮、分离、浓缩并干燥制得的,为衡量中药配方颗粒与临床汤剂是否基本一致的标准参照物,目前指纹图谱作为衡量中药材、标准汤剂、配方颗粒的综合有效手段[5]。
本实验采用HPLC法建立刺五加标准汤剂的指纹图谱,采用二极管阵列检测器(DAD)进行200~400 nm扫描,将各波长下的色谱图进行比较分析,结果发现在检测波长220 nm处,色谱信息丰富,基线稳定,故波长选择220 nm为检测波长。采用乙腈-水、甲醇-水、乙腈-0.2%磷酸进行梯度洗脱,结果表明,以乙腈-0.2%磷酸系统梯度洗脱对刺五加标准汤剂的分离效果较好。测定16批标准汤剂中有7个共有峰,并指认出原儿茶酸、紫丁香苷、绿原酸3种成分,16批标准汤剂指纹图谱相似度均在0.9以上,说明其质量稳定性较好,同时对其方法学进行了考察,结果均良好,说明此方法确定可行。
聚类分析法可将不同产地品质较接近的药材进行聚类,直观地表现药材间的亲疏关系及产地信息,并对它们进行归类,是一种很好的评价中药饮片质量的统计方法[8]。当类间的距离在10~15之间时,黑龙江伊春、吉林通化与黑龙江哈尔滨的刺五加亲缘关系较近聚为一类,吉林安图聚为一类,辽宁沈阳聚为一类,剩余其他样本聚为一类。主成分分析确定了3个主成分,累计方差贡献率67.649%,其中原儿茶酸、紫丁香苷、绿原酸对3个主成分贡献较大,这与文献报道基本一致[9-10]。按照综合得分计算吉林安图得分最高,说明质量最好。
综上所述,本研究应用指纹图谱和指标成分紫丁香苷含量测定相结合,初步建立了刺五加标准汤剂的质量评价方法。采用聚类分析和主成分分析法,将样本来源进行归类以及主要成分进行分析确定其主成分,从整体定性和指标成分定量的角度为刺五加相关制剂的制备和质量控制提供参考依据。
猜你喜欢
基层中医药(2022年5期)2022-10-24 01:27:40
今日农业(2022年15期)2022-09-20 06:54:16
中国药学药品知识仓库(2022年5期)2022-04-11 21:25:52
雪豆月读·低年级(2021年6期)2021-09-10 07:22:44
中国民间疗法(2021年14期)2021-08-30 08:24:56
北方音乐(2017年1期)2017-03-02 05:14:34
小学生作文选刊·低年级版(2016年7期)2016-08-06 13:09:32
中成药(2016年4期)2016-05-17 06:07:59
小学生·多元智能大王(2014年2期)2014-03-21 23:36:45
中国中医药现代远程教育(2014年18期)2014-03-01 04:30:05
