
进行性家族性肝内胆汁淤积症3型临床病理特征分析
2022-02-12 03:15翁宇航熊清芳刘杜先张胥磊杨永峰
临床肝胆病杂志 2022年1期
翁宇航, 熊清芳, 刘杜先, 张胥磊, 杨永峰
南京中医药大学附属南京医院(南京市第二医院) 肝病科, 南京 210003
进行性家族性肝内胆汁淤积症(progressive familial intrahepatic cholestasis,PFIC)是由基因缺陷引起的肝内胆汁淤积性疾病,因发病率极低,其流行病学、发病机制、临床特征、诊断、治疗、预后等方面的研究尚不充分[1]。PFIC共有6种临床类型,其中1~3型居多,国外文献报道1型和2型以婴幼儿期发病为主,3型以青少年或成人发病为主[2]。PFIC 3型(PFIC3)发病是由于ABCB4(ATP-binding cassette B4)基因突变,其编码的多耐药蛋白3(multidrug resistance protein 3,MDR3)表达下调或功能障碍所致[3],该基因突变还可引起妊娠期肝内胆汁淤积症(intrahepatic cholestasis of pregnancy,ICP)及低磷脂相关性胆石症(low phospholipid-associated cholelithiasis,LPAC)。考虑到PFIC3为罕见病,且目前可用的临床资料较少。本文回顾性分析PFIC3患者临床病理特征,以丰富国内PFIC3临床资料。
1 资料与方法
1.1 研究对象 收集2017年1月—2019年12月于本院就诊的1326例不明原因肝病患者的临床资料。不明原因肝病指无诱因持续或反复肝功能生化指标异常,或肝脏影像学检查异常6个月以上,通过常规病史询问、体格检查和实验室检查仍不能明确病因的肝病[4]。
1.2 纳入与排除标准 所有不明原因肝病患者均住院行肝组织穿刺,最终有218例患者在肝组织病理检查后仍不能明确病因的行二代测序全外显子检查。基因组DNA由EDTA抗凝的外周静脉血细胞获得,ABCB4突变检测采用遗传性胆汁淤积相关基因的Panel进行二代测序[5],对发现的突变位点进行Sanger测序验证[6]。诊断标准中,胆汁淤滞持续超过6个月考虑慢性胆汁淤积[7],肝硬化诊断参考2019年《肝硬化诊治指南》[8]。PFIC3目前尚无明确的诊断标准,诊断依据临床表现、检验、影像、肝组织学及基因检查。临床表现为慢性胆汁淤积,基因检测发现ABCB4致病突变的患者考虑PFIC3[9]。
1.3 资料收集 最终纳入PFIC3患者8例,其中1例因禁忌证未行肝组织穿刺,后经二代测序确诊为PFIC3。收集所有PFIC3患者的一般情况、临床症状体征、化验、影像、肝穿刺病理检查等资料进行分析。
1.4 肝组织活检 常规肝组织针刺活检,取长1.5~2 cm,直径0.1~0.2 cm肝组织,10%福尔马林固定、酒精脱水、石蜡包埋、HE染色,CK7免疫组化、目的蛋白用抗ABCB4(克隆P3Ⅱ-26,Thermo)对4 mm厚的石蜡切片孵育,后用多聚辣根过氧化物酶抗小鼠IgG,以二氨基联苯胺显色底物,并以正常肝和ABCB4缺乏症肝组织切片作为阴性和阳性对照。炎症分级(G)和纤维化分期(S)评分采用朔伊尔评分[10]。胆管损伤指小叶间胆管炎症浸润或胆管上皮损伤,包括胆管细胞肿胀、坏死、萎缩、退化等[11];胆管减少是10个以上完整汇管区中,20%~50%肝动脉无小叶间胆管伴行[12];胆管缺失是10个以上完整汇管区中,超过50%肝动脉无小叶间胆管伴行[13]。病理切片由临床医生和病理医师共同读片判定。
1.5 文献回顾 检索PubMed数据库,检索表达式为:(ABCB4[TI] OR MDR3[TI] OR Progressive familial intrahepatic cholestasis type 3[TI]) AND (histolog* OR patholog* OR Histopath* OR biops*),在检索结果中排除综述和非临床研究文献,最终纳入含有ABCB4病理描述和/或MDR3免疫组化的临床文献,对所有确诊为LPAC、 ICP、PFIC3患者的病理描述进行分析。
1.6 伦理学审查 本研究经南京市第二医院伦理委员会批准,批号: 2021-LY-kt052。
2 结果
2.1 一般资料 8例确诊PFIC3均为成年患者,其中男性占62.5%(5/8),女性占37.5%(3/8)。年龄最小18岁,最大54岁,中位年龄29.5岁。
2.2 临床表现 本研究队列中,50%(4/8)病例表现为慢性胆汁淤积;50%(4/8)病例表现胆汁性肝硬化,肝硬化病例中75%(3/4)合并显著的门静脉高压表现(表1)。
2.3 实验室检查 实验室数据均取自患者入院以来的极值(最小值~最大值)。所有患者肝功能指标均异常,ALT、AST均有不同程度地升高,87.5%(7/8)患者TBil升高,25%(2/8)患者出现低白蛋白血症,胆汁淤积相关指标明显升高,75%(6/8)患者ALP升高,100%(8/8)患者GGT升高,指标中以GGT升高最为明显;25%(2/8)患者表现为红细胞(RBC)降低,12.5%(1/8)患者白细胞(WBC)增高,25%(2/8)患者表现为凝血功能异常(表2)。

表1 8例PFIC3患者临床特征

表2 8例PFIC3患者实验室检查
2.4 影像学检查 50%(4/8)患者合并胆囊炎,25%(2/8)患者合并胆囊结石,25%(2/8)患者胆管扩张,75%(6/8)患者脾脏肿大,25%(2/8)患者出现肝硬化表现(表1)。
2.5 病理表现 PFIC3患者肝穿刺病理表现见表3和图1。7例患者均表现程度不同的炎症及纤维化,炎症以汇管区为主,3例(42.9%)伴轻度界面炎(G2)。纤维增生以汇管区为主,4例(57.1%)可见汇管-汇管为主的桥接纤维化(S3),其中3例(42.9%)可见不同程度的假小叶形成(S4)。胆管损伤是突出的病理改变,7例患者均可见不同程度胆管病变,表现胆管上皮炎细胞浸润、胆管上皮细胞缺失、排列紊乱等胆管损伤表现;3例(42.9%)表现小叶间胆管数量减少,其他4例(57.1%)表现小叶间胆管缺失。胆管缺失病例CK7免疫组化可见汇管区周围肝细胞阳性,提示肝细胞淤胆。MDR3免疫组化3例(42.9%)呈基本正常的肝细胞毛细胆管面表达,4例(57.1%)MDR3表达减少。
2.6 ABCB4突变检测 采用遗传性胆汁淤积相关基因的Panel进行二代测序,8例PFIC3患者均检测到ABCB4基因突变。5例检测到1个突变,为杂合突变;3例检测到2个突变,为复合杂合突变。ABCB4突变位点文献[14-16]报道有1个为已知变异,余下6个未知变异通过生物信息学软件分析提示可能致病(表1)。目前正在做体外功能验证,磷脂分泌初步实验结果提示也是致病的,因内容较多在此不具体叙述。

表3 PFIC3患者组织病理学及免疫组化染色特征
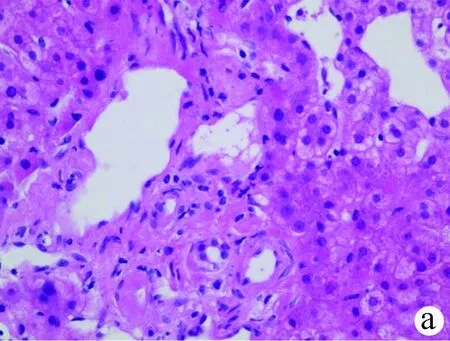
注:a,病例2汇管区小叶间胆管可见上皮细胞缺失,胆管损伤(HE染色,×400);b,病例4汇管区纤维增生,未见和小叶间动脉伴行的小叶间胆管,胆管缺失(HE染色,×200);c,病例1以汇管区为中心的纤维增生形成假小叶(Masson染色,×100);d,病例7 显示一个较大的汇管区胆管缺失,界面处可见细胆管,部分肝细胞CK7表达(免疫组化,×200);e,病例2 MDR3沿肝细胞毛细胆管面表达,表达量基本正常(免疫组化,×400);f,病例6约20%肝细胞毛细胆管面MDR3表达,表达明显减少,部分肝细胞浆MDR3弱表达(免疫组化,×400)。图1 PFIC3患者肝组织病理表现
2.7 ABCB4相关疾病病理文献回顾 在PubMed数据库检索,按照本文1.5表达式检索,其中符合表达式的文献共有98篇。排除综述和非临床研究文献,纳入含有病理胆管描述和/或MDR3免疫组化的文献17篇。病理描述文献中,7例LPAC胆管正常占71.4%(5/7),胆管减少占14.3%(1/7),胆管缺失占14.3%(1/7);6例ICP胆管正常占16.7%(1/6),胆管减少占50%(3/6),胆管缺失占33.3%(2/6);8例PFIC3胆管减少占25%(2/8),胆管缺失占75%(6/8)。MDR3免疫组化文献中1例LPAC患者为正常表达;21例PFIC3正常表达占9.5%(2/21),表达减少患者占23.8%(5/21),表达缺失占66.7%(14/21)。病理文献结果详见表4。
3 讨论
PFIC是一组罕见疾病,通常发生在婴儿期和儿童期,目前已报道6种类型,其中ABCB4基因缺陷可导致PFIC3。ABCB4基因编码磷脂转运蛋白MDR3,该基因缺陷可以引起MDR3转运障碍,磷脂不能被转运到胆汁中。生理情况下,胆汁中磷脂、胆盐、胆固醇比例适中。当磷脂比例减少时,胆盐和胆固醇占比升高,胆盐破坏胆管,胆固醇淤积从而致病[34]。ABCB4基因突变还可以引起ICP及LPAC等疾病,但严重程度相比PFIC3较轻。
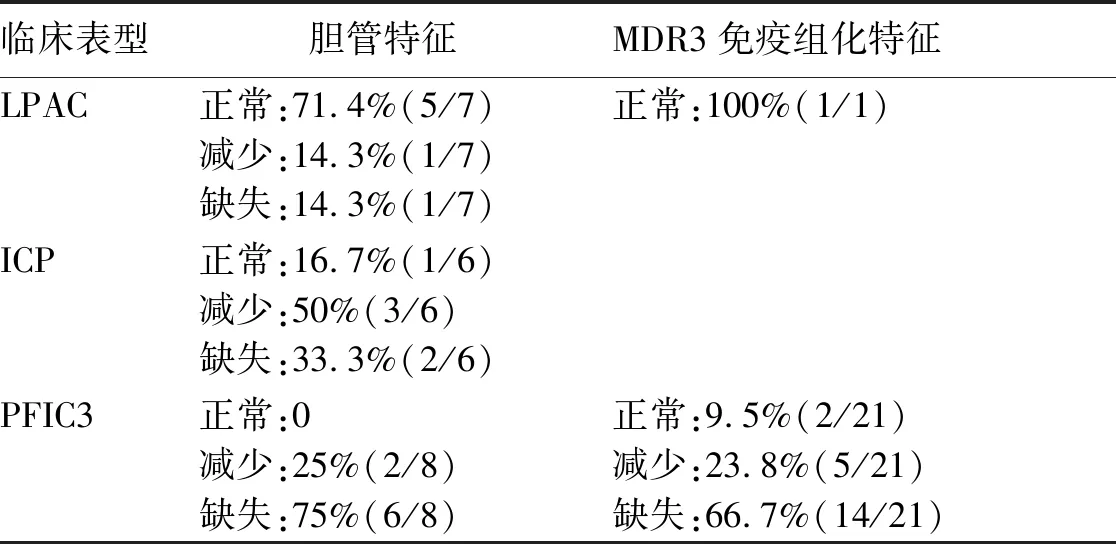
表4 ABCB4突变患者病理特征文献[17-33]回顾
PFIC3目前在国内流行状况不明。患者通常在婴儿晚期(约三分之一的病例)至青春期出现胆汁淤积,轻者表现为无黄疸的慢性胆汁淤积症,重者可进展至门静脉高压、肝硬化和肝衰竭。由于该病起病隐匿,肝硬化和消化道出血可能是儿童或成年阶段首发症状。患者发生胆固醇结石的风险也有所增加[35]。体征包括黄疸、肝肿大。本队列中8例患者占同期不明原因肝病患者的0.6%(8/1326),中位年龄为29.5岁,患者均表现为长期慢性胆汁淤积,严重者表现胆汁性肝硬化,可出现黄疸、门静脉高压、食管胃静脉曲张出血等症状,符合文献报道结果。患者发病年龄及疾病严重程度各异,可能是突变致病性不同或与个体差异性有关。
PFIC3患者实验室检查以胆汁淤积为主,通常GGT和ALP增高[36],可伴有氨基转移酶等炎症指标增高。当出现肝硬化时,可出现黄疸、低白蛋白血症、凝血功能异常[9]。本队列患者均有明显的胆汁淤积,表现为胆酶(GGT、ALP)显著升高,其中GGT升高明显;其他亚型(1型、2型、4型、5型、6型)家族性胆汁淤积症均表现低GGT胆汁淤积,有助于临床鉴别。患者1、6有肝硬化表现,如凝血功能异常及低白蛋白血症。
影像方面超声是一项最基本的检查,除了部分PFIC3病例存在胆结石外,它还有助于排除其他梗阻性原因引起的胆汁淤积。磁共振胆道成像可清晰显示胆管结构,有助于本病鉴别诊断。本队列中,患者多伴有胆囊炎、胆囊结石、胆管扩张,这符合PFIC3的病理生理学特点。一方面胆固醇等物质析出容易造成结石;另一方面,结石以及反复的胆管损伤可能造成胆管、胆囊炎症,进而胆管扩张,严重者也可有胆汁性肝硬化。
肝组织学对诊断非常重要。在PFIC3中,组织学表现除炎症及纤维化等非特异性病理改变外[37],胆管损伤是该病的主要病理特征,且随病情加重,可出现胆管缺失[38-39]。根据文献回顾结果,ICP及LPAC病理多表现为胆管正常或减少,而PFIC3多表现为胆管缺失,缺失数量较多者往往预后较差,此类患者多表现肝硬化或在早期行肝移植治疗。以上结果提示临床表型越重,胆管缺失比例也越高。本队列所有病例均表现胆管损伤伴胆管减少或胆管缺失,且纤维分期S3以上病例均表现胆管缺失,提示胆管损伤程度与疾病严重程度有关。MDR3表达在PFIC3中可以表现为正常、减少或缺失[40-41]。虽然MDR3表达减少或缺失有助于诊断,但正常表达并不排除PFIC3的诊断。因为突变可能导致MDR3功能丧失,但仍可以正常合成和定位[42],本队列中57.1%(4/7)病例MDR3表达减少,42.9%(3/7)病例MDR3表达正常,且MDR3表达和病情轻重无相关性。因此MDR3免疫组化结果正常时仍不能排除诊断,必要时需基因检测进一步确认。
总之,不明原因慢性或反复伴GGT升高的胆汁淤积的患者,需考虑PFIC3可能;LPAC病史和家族史支持本病诊断,超声、磁共振、CT等影像检查有助于鉴别诊断和病情评估;胆管损伤/缺失伴不同程度的炎症、纤维化是该病的病理特征;MDR3免疫组化显示表达减少或缺失有助于诊断,但MDR3表达正常也不能排除该病;该病的确诊仍有赖于基因检测,加强临床和基因检测实验室之间的沟通至关重要。由于PFIC3发病罕见,未来需多中心的临床队列研究和基础研究,以进一步明确该病的临床特征、诊断和治疗方案。
利益冲突声明:本研究不存在研究者、伦理委员会成员、受试者监护人以及与公开研究成果有关的利益冲突。
作者贡献声明:翁宇航负责收集数据,资料分析,撰写论文;熊清芳、刘杜先、张胥磊指导撰写文章;杨永峰负责课题设计,拟定写作思路,修改论文并最后定稿。
猜你喜欢
基层中医药(2022年7期)2022-11-17
珠江水运(2022年17期)2022-09-25
传染病信息(2022年2期)2022-07-15
中国典型病例大全(2022年12期)2022-05-13
智慧医学(2021年1期)2021-09-10
珠江水运(2021年15期)2021-08-29
医学食疗与健康(2021年27期)2021-05-13
学习与科普(2019年15期)2019-09-10
水能经济(2017年6期)2017-10-19
中国民族民间医药·下半月(2016年8期)2016-10-24
