
转化生长因子-β亚型在慢性阻塞性肺疾病急性加重期患者中的表达研究*
2022-02-01 10:40江山,袁竞,秦超,戴曦
黑龙江医药 2022年24期
江 山,袁 竞,秦 超,戴 曦
1.都江堰市人民医院呼吸与危重医学科,四川 成都 611830;2.泸州市人民医院检验科,四川 泸州 646000;3.西南医科大学附属医院呼吸与危重症医学科,四川 泸州 646000
慢性阻塞性肺疾病(COPD)是一种危害人类健康的慢性气道性疾病,此疾病重要特点为进行性加重和不可逆地呼气气流受限。此疾病发病率高,预测未来数十年里此疾病的发病率及死亡率仍然会持续增加,它可以使患者生活质量出现严重下降、劳动能力缺失[1]。有害刺激物吸入导致气道内的多种细胞功能失调,释放炎症介质,趋化炎症细胞,使气道黏液分泌过度、细胞外基质病理性沉积,从而导致慢性气道炎症引发气道重塑以及管腔狭窄。黏液高分泌与气道重塑这两个原因是导致慢阻肺发生发展的关键病理特征,而肺泡巨噬细胞(AM)和中性粒细胞则是导致COPD 炎症的最重要两大类炎性细胞。目前国内关于AM 在COPD 发生发展中的机制研究尚不完善,发生急性加重的机制亦不完全清楚。目前有部分研究发现TGF-β/Smad信号传导通路与气道重塑关系密切,而其中转化生长因子-β1 已被大家确认为一种强致纤剂,其表达水平与纤维化疾病的严重程度和死亡率明显正相关[2-3]。然而在人体中TGF-β 与COPD 关系的研究尚不明确,而TGF-β2 与成纤维细胞的关系目前仍然尚存在争议。本研究旨在探讨TGF-β1/β2/β3在COPD 发生发展的过程中发挥的作用,进一步研究三组转化因子与COPD 急性加重期关系及相关的作用机制,现将结果报告如下。
1 资料与方法
1.1 一般资料
按照2016GOLD 诊断标准,选取2019 年1 月—2021 年12 月西南医科大学附属就诊的慢阻肺急性加重期病患14例作为实验对象,门诊或者住院治疗的COPD 稳定期病患15 例,正常志愿者8 例,所有实验对象均排除外支气管哮喘、支气管扩张、糖尿病、高血压、肺癌等合并症。慢阻肺急性加重期组在行诱导痰检查前4 周内均未给予任何方式的激素治疗。1%二硫苏糖醇(DTT,Merk公司);Human TGF-β1/β2/β3 ELISA 试剂盒(诚林生物公司);兔抗人Smad2/3抗体、荧光标记人抗兔IgG 抗体(Santa Cruz);超声雾化器;激光共聚焦显微镜;酶标仪;MASSON 复合染色液、亮绿染色液等。
1.2 主要实验方法
生理盐水充分漱口后,分别用3%、4%、5%浓度梯度的NaCL 溶液进行超声雾化,5 分钟/次,总雾化的时间不大于30分钟,有咳痰时再次用生理盐水漱口,继而指导患者深咳,将深部的痰液收集至无菌痰杯内密封保存,每份痰液标本量大于等于2 mL。同时进行电子支气管镜检查并采集研究对象气道黏膜上皮活检标本6 份,其中三份立即予以甲醛固定保存,另外三份放至锡箔纸4 ℃保存,当日提取蛋白。标本处理,挑取痰液标本样品进行涂片染色并在显微镜下观察,如若显微镜下每低倍镜白细胞数大于等于25 个,或白细胞在10~25 个/Lp 并且上皮细胞数目小于10个,研究者则判定其为标准的合格的标本样品。挑取没有唾液附着的痰液栓,加入0.1%二硫苏糖醇(DTT)进行充分溶解后,加入适量的磷酸盐缓冲溶液(PBS)进行稀释后离心。(1)收集上清液,采取ELISA 法检测其中TGF-β1/β2/β3 的浓度。(2)沉淀的细胞成分分别以0.4%台盼蓝染色并在血细胞计数板下统计成活细胞率及细胞总数,并行瑞氏染色后行细胞分类并进行计数,HE 染色并鉴定AM并调整其浓度,将悬液于37 ℃,5%CO2环境中培养2 h,取出细胞爬片,采用免疫荧光法检测AM 中Smad2/3。(3)气道黏膜活检组织以石蜡切片采取常规方式脱蜡后,先后以MASSON 复合染色液、亮绿染色液染色5 min,给予常规的脱水,透明,然后进行封片处理,进而在镜下观察气道上皮组织胶原纤维增生情况。(4)锡箔纸保存气道粘膜活检组织,提取总蛋白并测定蛋白浓度,配制SDSPAGE 凝胶,按步骤完成电泳、转膜、封闭、一抗、二抗温育、洗膜、显影、测量灰度值。
1.3 统计学方法
采用SPSS 19.0 软件进行统计分析。计量资料以均数±标准差(±s)表示,组间比较采用t检验。计数资料以例数和百分比(%)表示,组间比较采用χ2检验。以P<0.05为差异有统计学意义。
2 结果
2.1 一般资料
COPD 急性加重期组的平均年龄为(66.79±6.34)岁,COPD 稳定期组为(64.73±5.99)岁,正常对照组的平均年龄为(χ2=1.975,61.25±6.73)岁,所有三组实验对象的年龄,差异无统计意义(P>0.05);三组的男女比分别为7/7、8/7、4/4,构成比,差异无统计学意义(F=6.667,P>0.05)。三组的FEV1%预计值明显不同,分别为48.88±7.98、59.11±6.98 和95.25±5.75,差异有统计学意义(χ2=110.708,P<0.05)。
2.2 诱导痰炎细胞分类计数
慢阻肺急性加重期组诱导痰中炎症细胞数最多并且以中性粒细胞为主,慢阻肺稳定期组高于对照组诱导痰的炎细胞总数。慢阻肺患者不同病理阶段诱导痰中AM 数目明显有较大的差异,慢阻肺急性加重期组AM 计数最高,见表1、表2,图1。

图1 (A)诱导痰中炎细胞及AM计数,(B)诱导痰中炎细胞分类计数

表1 实验对象诱导痰炎细胞总数、分类计数
2.3 诱导痰TGF-β亚型表达水平及巨噬细胞中Smad2/3荧光强度
分别检测诱导痰上清液中TGF-β1/β2/β3 的含量并进行比较后发现,慢阻肺患者上述三个指标的表达水平明显高于对照组,并且不同病程时期诱导痰中TGF-β1/β2/β3表达水平不同,慢阻肺急性加重期组中含量高于慢阻肺稳定期组,差异有统计学意义(P<0.05),见表2,图2A。慢阻肺患者不同病理阶段诱导痰中Smad2/3 表达水平有明显且较大的差异,慢阻肺急性加重期组巨噬细胞中Smad2/3 荧光强度最高,慢阻肺稳定期组巨噬细胞Smad2/3 荧光强度亦高于对照组,见表2,图2B、C。
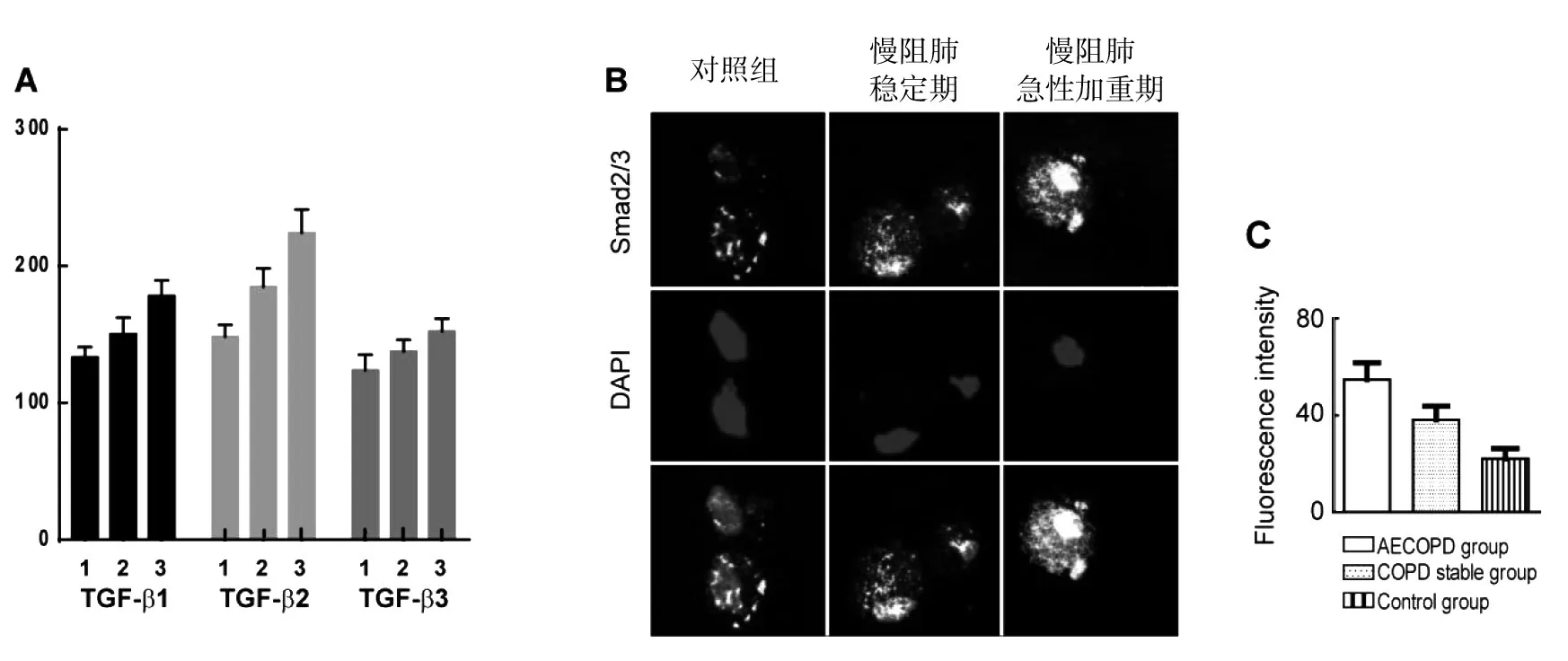
图2 (A)诱导痰中TGF-β1/β2/β3的表达水平,(B、C)AM中Smad2/3表达强度检测

表2 各组诱导痰中TGF-β表达水平以及Smad2/3荧光强度
2.4 TGF-β 亚型与慢阻肺患者肺功能以及AM 数目和Smad2/3的相关性分析
根据慢阻肺患者总体相关性分析,TGF-β1/β2 在患者诱导痰中的表达水平与患者肺功能显著负相关,与痰中AM 数目和Smad2/3 荧光强度之间均呈显著正相关,但TGF-β3与肺功能、AM数目均没有相关性,见图3。
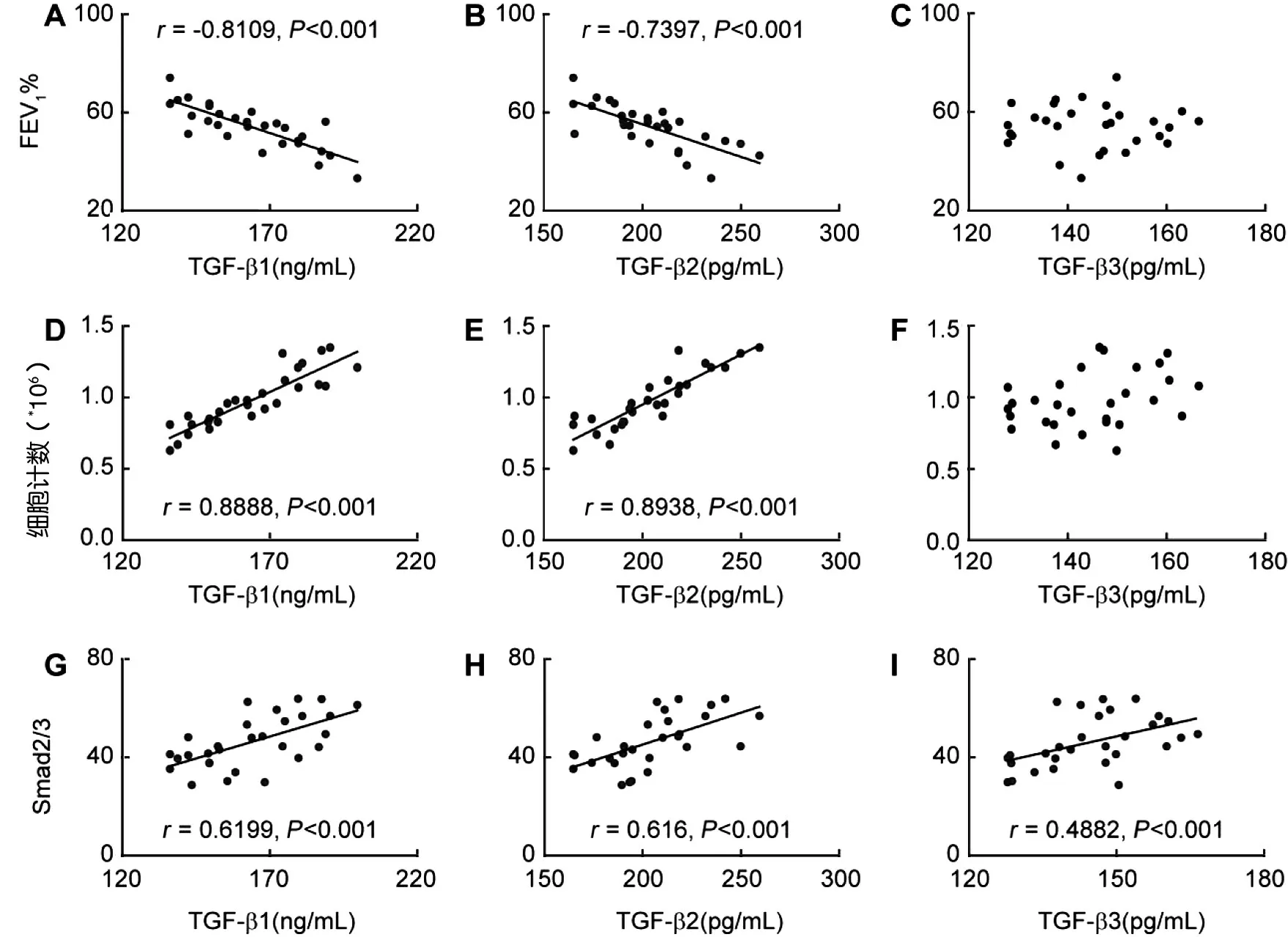
图3 TGF-β1/β2/β3表达水平与患者肺功能巨噬细胞数目和Smad2/3的相关性分析
3 讨论
炎症细胞机制在目前的研究中认为其是COPD 的重要病生机制之一[4-5]。本实验选择主要来自周边气道小分支的诱导痰作为实验对象,其细胞及生化成分更加集中。细菌、病毒包括支原体衣原体均导致COPD 的急性加重,细菌、病毒等病原体本身并不直接导致TGF-β的改变,而是会通过炎症细胞经过一系列通路来影响TGF-β 的表达水平。与实验结果相一致,中性粒细胞在慢阻肺急性加重期患者和COPD 稳定患者中都是最为重要的炎症细胞,与Barnes[6]的研究结果类似,中性粒细胞在急性加重期中持续稳定的存在,而且在稳定期气道炎症中仍旧持续稳定的存在。诱导痰中性粒细胞数目可预测COPD 的严重程度。中性粒细胞是IL-8的主要来源,具有通过其合成并释放丝氨酸蛋白酶和氧化因子诱导组织损伤的能力,气道中性粒细胞数量的增加与气道气流限制程度密切有关[7]。同时研究者发现COPD 急性加重期组嗜酸性粒细胞比率低于稳定期组,并发现对照组及COPD 稳定期组嗜酸性粒细胞率均不低于2%,与Hall[8]报道的COPD 加重期患者痰中的嗜酸性粒细胞明显增加,吸入激素后痰中的嗜酸性粒细胞比例降低的结果存在不一致。
AM 定位于肺泡壁,同时具有抗炎以及促炎的双向效应,被认为是另一种主要的气道炎症细胞,多个实验研究认为AM 导致了气道重塑和肺功能损伤,其数量与肺实质的损害严重程度正性相关。人体中的巨噬细胞均可被激活并释放大量的炎症介质、酶和细胞因子,包括IL-8和白三烯,TNF-α、NF-κB 等,过往的实验及临床研究认为AM通过炎症介质的相互作用可以导致气道杯状细胞增殖和胶原纤维增生,进而导致黏液高分泌与气道重塑;另一方面,AM 能分泌中性粒细胞趋化因子,在慢阻肺中促进毛细支气管慢性炎症,分泌弹性蛋白酶破坏蛋白酶抗性蛋白酶系统的平衡。而上述炎症效应在慢阻肺急性加重期中被放大[8]。
然而在本实验中通过研究发现,除了以上机制,AM可能参与诱导的TGF-β/Smad2/3 蛋白信号通路或在慢阻肺病程进展中发挥了重要作用。目前发现TGF 至少有6 个亚型,但是在人类中仅发现3 个同分异构体的亚型,分别为TGF-β1/β2/β3,在哺乳动物体内几乎被所有细胞分泌,并广泛参与机体损伤修复、胚胎发育、细胞增殖分化、细胞外基质代谢、炎症、免疫等方面。TGF-β1/β2/β3 是以复合体形式分泌,三者之间相互关联影响而发挥自身的调节、周边的调节功能,也能通过血液、淋巴液循环对远处的靶细胞其相应的作业。多个证据显示TGF-β1 的表达主要归因于巨噬细胞。TGF-β1 被认为是目前最关键的致纤维化因子和最强的细胞外基质沉积的促进剂,一方面可促进细胞组织修复的同时也在促进纤维化的发生由此引起气道重塑使气体流速受限,另一方面能延长Th2 细胞的生存而使IL-13 生成增加,进一步的刺激TGF-β2 产生及释放,以没有特异性方式诱发和导致气道上皮黏蛋白的过度表达[3]。与此同时它还可以促进炎性介质的释放,通过一级一级的放大加重炎症的反应,从而导致细胞外间质代谢失衡、引发成纤维细胞激活、积聚、杯状细胞的增生、促进黏液高分泌。
在以往的多个研究中已经发现TGF-β 是通过Smad 蛋白通路从而完成其信号转导的。Smads 蛋白家族中Smad2/3/4/7 可以参与TGF-β 信号传导,也是到今天所知晓的细胞内唯一的TGF-β受体作用的底物。未活化的受体调节型Smad 蛋白的MH1 区及MH2 区相互抑制,发挥不了其生物学效应,经活化后的R-Smad 蛋白其MH1 区的核酸定位样序列使Smad 蛋白向细胞核转移,并与相应的靶基因结合,同时需要Co-Smads作为中间媒介并保持Smads低聚体的构型。TGF-β1/β2/β3 通过该通路,由配体结合到细胞表面的TβRII,再激活TβRI 的丝氨酸/苏氨酸激酶区进一步的活化TβRI,形成三聚体,TβRI 再此激活Smad2/3 从而使其磷酸化进入细胞核内,将信号转导入细胞核,进而引起气道黏膜上皮内胶原纤维增生,气道重塑,出现不可以逆转的气体流速受限。有的研究得出气道重塑仅有TGF-β1作用其中而并不包括其他TGF亚型,有研究认为[9],TGF-β3可以促使肺内纤维化组织修复,在修复的同一时间下调TGF-β1 诱导的基因表达,TGF-β3 可以抑制炎症引发的TNF-α 水平上调,从而减轻炎症的程度。TGF-β2 则被单独发现有助于增加嗜酸性粒细胞、淋巴细胞和胶原沉积。
本实验中研究者发现,慢阻肺患者气道黏膜存在程度不一的胶原纤维过度沉积,伴有黏液高分泌,这也是引起慢阻肺急性加重期发生发展的关键病生基础。TGF-β1/β2/β3 过表达在慢阻肺病程中贯穿始终,不能证明TGF-β3 的保护性作用。通过对诱导痰研究,我们证实了AM 在慢阻肺急性加重期中数量增多且高表达Smad2/3 蛋白,同时TGF-β1/β2 高表达且与AM 数量、Smad2/3 蛋白显著正相关,可以认为AM 导致TGF-β高表达并激活TGF-β/Smad2/3 蛋白信号通路,引起气道慢性炎症和广泛小气道胶原纤维沉积,从而导致器质性气流阻塞、肺功能恶化,在气道重塑中均起着至关重要的作用,也是导致急性加重的重要且关键的因素之一。通过检测患者诱导痰中TGF-β1/β2的水平高低可以作反映慢阻肺的严重程度、并对病情的检测、疗效的观察以及转归的判断起到一定的辅助作用,但其具体的通路及作用方式仍需进一步研究。
猜你喜欢
基层中医药(2022年5期)2022-10-24
中国药学药品知识仓库(2022年10期)2022-05-29
现代临床医学(2021年6期)2021-11-20
中国慈善家(2021年5期)2021-11-19
英语文摘(2019年6期)2019-09-18
商周刊(2018年11期)2018-06-13
医学研究杂志(2015年3期)2015-06-10
中国医疗美容(2015年4期)2015-04-27
小说月刊(2015年3期)2015-04-19
中国当代医药(2015年21期)2015-03-01
