
儿童髓母细胞瘤分子病理学分型与诊断及预后的研究进展
2022-01-28 09:28高鑫义邹有瑞
中国实验诊断学 2022年1期
高鑫义,邹有瑞,黄 灵,马 辉*
(1.宁夏医科大学,宁夏 银川750004;2.宁夏医科大学总医院 神经外科,宁夏 银川750004)
髓母细胞瘤(medulloblastoma,MB)是由Bailey与Cushing于1925年最先提出的,是一种可发生在颅脑的任何部位,但绝大多数生长在第四脑室顶之上小脑蚓部的胚胎源性肿瘤[1]。世界卫生组织(World Health Organization,WHO)在中枢神经系统肿瘤分级中将其分为WHO IV级,表明其在组织学上具有高度恶性,其来源可能与生命早期各种离散的神经干细胞或神经祖细胞群未继续分化相关[2]。MB是儿童CNS最常见的恶性肿瘤,约占所有儿童CNS肿瘤的15%-20%[3]。虽然MB诊断的高峰年龄约为 6-8岁,但其也可能发生于婴儿阶段及成人时期,同时在部分亚型中可呈双峰样特点[4-5]。在治疗方案上,MB目前采用最大安全范围切除术联合化疗和全脑全脊髓放射治疗(craniospinal irradiation,CSI)等综合治疗方案,可极大地提高患者的生存率,但仍约30%的患者最终死于这种疾病[2,6-7]。根据组织学分类,2007年WHO系统地将MB分为4种病理亚型:经典型 (classic) 、促结缔组织增生/结节型( desmoplastic/nodular,DN) 、广泛结节型(medulloblastoma with extensive nodularity,MBEN) 和大细胞/间变型(large cell/anaplastic,LC/A)[8]。随着分子生物学的发展,2016年WHO将CNS肿瘤的分类进一步完善,根据MB分子生物学特性引入了4种亚型,分别为WNT型MB、SHH型MB、3型MB和4型MB[5,9]。进一步的研究表明,根据基因组分析,可将这些亚型分为更详细的亚组[10-12]。目前,DNA甲基化图谱测定MB分子病理在国际上被推荐作为MB分子分型诊断的金标准,在一定程度上取代了基因表达图谱[2,13-14]。现对MB分子病理学分型特点及其诊断和预后作一综述。
1 髓母细胞瘤分子分型的特点
1.1 WNT型MB
WNT型MB以其发病机制中起重要作用的WNT信号通路而命名,约占所有MB的10%[15]。这种MB在青少年中最为常见,而在婴幼儿中非常罕见,男女性别比相近。在组织学水平上,几乎所有病例均为经典型,极少部分病例可为LC/A型[16]。该亚型中约有85%-90%存在CTNNB1基因的第3号外显子活化突变,可作为该亚型最显著的标志,其中β-catenin蛋白是WNT信号通路重要的效应因子[2]。β-catenin蛋白在细胞核内聚集,与TCF-LEF家族转录因子的发生共同作用与WNT型MB的发生密切相关[17-18]。其他常见的突变包括DDX3X(36%)、SMARCA4(也称为BRG1,19%)、TP53(14%),同样也包括CSNK2B(14%)、PIK3CA(11%)、EPH47(8%)这类在临床上可采取一定措施控制的突变[2]。6号染色体出现单倍体是WNT型MB在细胞遗传学上的较为显著的特征,同时常伴有CTNNB1基因突变,临床上,二者中任意为阳性,可作为WNT型MB的判定标准。因此,可将CTNNB1基因中β-catenin蛋白作为其分子病理诊断标准。然而6号染色体出现单倍体和(或)CTNNB1基因突变并非存在于所有的WNT型MB中,会存在10%-15%的真正WNT型MB患者被漏诊[19]。Clifford等[20]用 SNF(similarity network fusion)的方法进一步鉴别出WNTα和WNTβ两个子亚型:WNTα主要分布在儿童群体,98%d的病例伴随6号染色体出现单倍体;WNTβ主要分布在青少年及成年人群体中,很少(29%)伴有6号染色体出现单倍体[20]。WNT型是所有MB亚型中预后最好的亚型,肿瘤的转移很少发生,16岁以下的患者5年生存率超过95%,这可能与缺乏完整的血脑屏障导致肿瘤内存在高浓度化疗药物的积累相关[21]。
1.2 SHH型MB
SHH型MB以其发病机制中起重要作用的SHH信号通路而命名,约占所有MB的30%。该型MB发病年龄呈双峰样特点,好发于婴幼儿和成人,男女性别比相近[9,22]。在组织水平上,它可以表达为四种病理类型中的任何一种,但几乎所有的MBEN都属于这种类型。目前研究表明,肿瘤细胞起源于小脑的颗粒神经元前体细胞(granule neuron precursors,GNPs),与其他亚型MB有所区别的是肿瘤好发于一侧的小脑半球[9,23],大多数肿瘤包含SHH信号通路相关基因生殖系或体细胞突变或拷贝数改变,致使SHH信号激活,推动肿瘤的进展。常见突变或缺失的基因包括PTCH1(43%)和SUFU(10%)的突变或缺失[24],还经常可观察到SMO(9%)的活化突变、GLI1或GLI2(9%)和MYCN(7%)的扩增[24]。此外,TP53信号通路(9.4%)和PI3K通路(10%)的反复突变也可能是该型肿瘤发生的关键驱动因素,83%的SHH型MB病例(包括几乎所有成人SHH型病例)中也发现TERT(telomerase reverse transcriptase)启动子突变,年龄分布差异明显[12,24]。同时,在该型MB中也发现了其他神经胶质瘤中常见的IDH1基因突变,其导致DNA超甲基化表型,类似于在其他IDH1/2突变型肿瘤中发现的表型[24]。该型MB的发生可能与某些肿瘤抑癌基因丢失相关,如PTCH1(位于9q22)和SUFU(位于10q24),它们分别编码SHH信号的负调节因子,以及其他潜在的调控因子,其染色体9q和10q的缺失可作为该型MB的细胞遗传学标志性特征[24]。Schwalbe等[10]研究发现可依据年龄及组织学、分子病理等特征将SHH型进一步分为MBSHH-Infant(<4.3岁)和MBSHH-Child (≥4.3岁):MBSHH-Infant组织病理分型多为DN型,富含SUFU突变,总体预后较MBSHH-Child型稍好,5年总生存率约为62%;MBSHH-Child组织病理多为LC/A型,TP53、TERT突变和MYCN、GLI2扩增,染色体9q缺失和9p获得是其分子生物学常见特征,5年总体生存率约为58%。Cavalli等[12]进一步将SHH型MB分为4个子亚型:SHHα、SHHβ、SHHγ和 SHHδ。SHHα主要发生于3-16岁儿童,预后最差,TP53突变,富含MYCN、GLI2扩增,其中TP53突变型因其极高风险,WHO将TP53突变型作为一个的单独的类别以判断预后;SHHβ多见于婴幼儿,多伴有转移,预后较差;SHHγ组织病理多为MBEN,预后较好;SHHδ主要由成年人组成,富含TERT启动子突变[12]。SHH型MB总体预后因临床(年龄和转移状态)和分子(MYCN扩增和TP53突变状态)特征而异,预后中等,5年生存率约在60%- 80%[12,25]。
1.3 3型MB
3型MB真正起源有待进一步研究,可能来源于神经干细胞群,发病部位常位于脑干附近的第四脑室[26-29]。这种类型的MB约占所有MB病例的四分之一,它通常发生在10岁以下的婴幼儿中,在成人群体中很少见,男性患者接近女性患者的2倍[9,22]。该型MB组织病理类型以经典型为主,同时大部分LC/A也属于此型[22]。MYC基因高水平扩增(17%)可作为该型最显著的特征,非编码RNA PVT1共扩增常伴随其发生,且后者可以协助MYC蛋白高水平稳定表达[2,24,30-31]。复发性体细胞突变在该型肿瘤中很少见,只有四种基因突变存在于超过5%的病例中:SMARCA(9%)、KBTBD4(6%)、CTDNEP1 (5%)和KMT2D(5%),此外还有MYCN(5%)和 OTX2(3%)的扩增,其中OTX2在控制不同祖细胞分化的过程中起着至关重要的作用[2,24,32]。研究表明,在15%-20%的MB中存在增强子劫持(enhancer hijacking)事件导致原癌基因GFI1和GFI1B表达上调可能是该亚型重要的驱动事件[33]。染色体1q、7和17q的扩增,10q、11q、16q和17p的缺失,以及等臂染色体17q(isochromsome17q,i17q)的存在是3型MB在细胞遗传学方面主要表现,同时i17p可能作为3型MB不良预后的重要标志[24,34-35]。NPR3蛋白可作为3型MB的分子病理诊断标准之一,但其免疫组化染色阳性能否作为3型MB的特征性标志物有待进一步研究[3,36]。Schwalbe等[10]将Group3型分为MBGrp3-HR和MBGrp3-LR:MBGrp3-HR和MBGrp3-LR组织病理分别以LC/A型和经典型最为常见。前者富含MYC扩增、GFI1突变、il7q,5年总生存率为37%;后者以婴幼儿患者居多,常伴多个染色体缺失,预后相对较好,5年总生存率为69%。Cavalli等[12]综合聚类分析结果鉴别Group3α、Group3β和Group3γ 3个子亚型:其中Group3α多伴有转移,而Group3β转移发生较少,但Group3α预后相对较好,以婴幼儿患者为主。3种子亚型中以Group3γ预后最差,以伴8q重复,富含i17q,MYC拷贝数增加为其显著特征。该型MB易发生转移(诊断时为40%-50%),临床预后在4种亚型中最差,5年生存率低于60%[9]。
1.4 4型MB
4型MB约占所有MB的35%-40%,好发于儿童和青少年,在男性中更为常见,男女性别比近 2∶1[9]。4型MB起源细胞尚未确定,疑似起源于胚胎上菱唇前体细胞,发病部位与3型MB相近,通常位于第四脑室[26,37-38]。在组织学上,以经典型最常见[22]。常见的体细胞突变在该亚群中也很少见。4型MB最显著的驱动事件为PRDM6活化过表达[39]。其他的基因突变还包括KDM6A(9%)、ZMYM3(6%)、KMT2C(6%)和KBTBD4(6%)的突变,以及MYCN(6%)和OTX2的扩增(6%)、CDK6(6%)、GFI1以及GFI1B过表达(5%-10%)[39]。其中,KCNA1可作为4型MB诊断的分子病理学依据[9]。Schwalbe等[10]以高风险和低风险为特征,将4型MB分为MBGrp4-HR和MBGrp4-LR,其中高风险组多有PRDM6的扩增,富含等染色体17q,10年生存率为36%;低风险组多有MYCN扩增,伴7号和17q染色体的扩增和8号、11号染色体的缺失,10年生存率约为72%,二者组织病理均多为经典型。Cavalli等[12]综合聚类分析进一步将4型MB分为Group4α、Group4β和 Group4γ三个子亚型:Group4α富含MYCN的扩增,伴染色体8p的缺失和7q的扩增;Group4β中SNCAIP扩增明显,伴随着广泛存在的i17q;Group3γ富含CDK6的扩增增加。虽然该亚型也经常发生转移(诊断时为35%-40%),但该亚组的总体生存结果是中等的,与SHH型MB相近,介于WNT型MB和3型MB之间,复发往往发生较晚[9,40]。
2 髓母细胞瘤危险分层的特点及预后
2.1 危险度分组[41]
目前国内根据年患者龄、手术切除程度、有无转移和组织病理类型将MB进行危险度分组:①对于年龄>3岁的儿童MB,标危的判定标准为:术后肿瘤残留病灶 ≤1.5 cm2,无扩散转移;高危的判定标准为:肿瘤残留病灶>1.5 cm2;有MB肿瘤转移(包括神经影像学证据、手术14天后腰穿或脑室脑脊液阳性细胞学证据或颅外转移);病理组织学为弥漫间变型。②对于年龄≤3岁儿童MB,标危的判定标准为:肿瘤残留病灶≤1.5 cm2、无扩散转移、病理组织学为为促结缔组织增生型或广泛结节型;高危组:除标危外全部判定为高危(表1)。
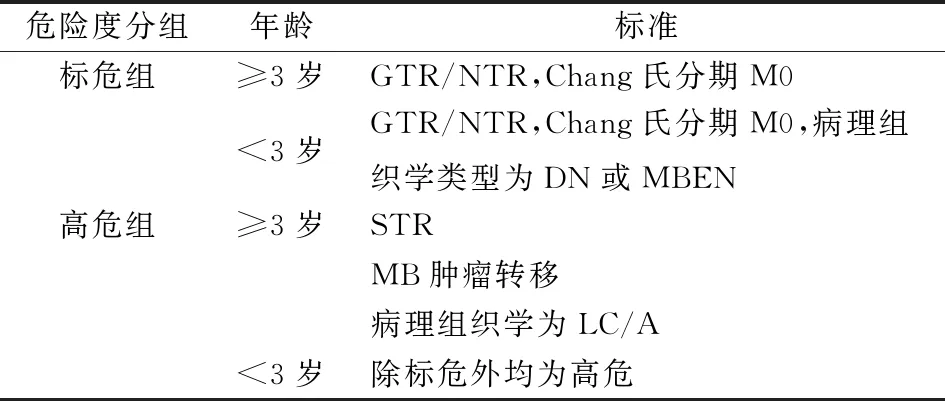
表1 危险度分组
2.2 分子危险度分层
2017年Schwalbe[10]等建立生存模型分析,根据5年生存率提出危险分层:①低危组(>90%):MBWNT,没有高危标志的MBSHH-Child(高危标志:伴有转移,术后残留病灶>1.5 cm2,大细胞型/间变型,MYCN扩增),没有MYC扩增和伴染色体13缺失的MBGrp3/Grp4;②标危组(75%-90%):没有 MYC 扩增的MBGrp3-LR/Grp4-LR;③高危组(40%-75%):没有MYC扩增的MBGrp3-HR/Grp4-HR;④超高危组(<40%):有高危标志的MBSHH-Child,MYC扩增的MBGrp3。
但目前我国乃至国际上仍主要根据2015年海德堡会议[2,41]结合分子分型进行危险度分层。分子危险度分层根据5年生存率划分为四个组:①低危组(>90%):无扩散转移且年龄<16岁的WNT型,无扩散、转移和伴11号染色体的缺失或17号染色体重复的4型MB;②标危组(75%-90%):无扩散转移、不伴TP53突变和MYCN扩增的SHH型,无MYC扩增的3型MB,无扩散转移和不伴染色体11的缺失的4型MB;③高危组 (50%-75%):伴有转移的TP53野生型SHH型,无扩散转移的MYCN扩增的SHH型,伴有转移的4型MB;④超高危组(<50%):伴有转移的MYC扩增的3型MB,伴有TP53突变的SHH型(表2)。
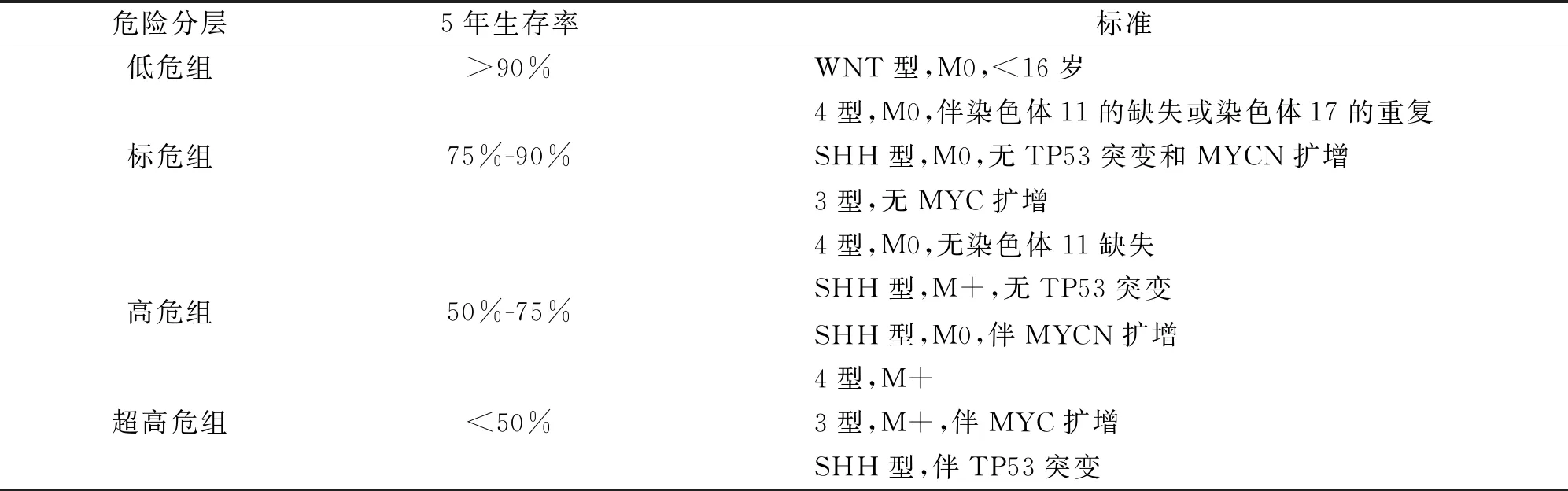
表2 分子危险度分层
3 总结与展望
MB具有高度异质性,虽然目前手术联合放疗、化疗的综合治疗方案可以有效地改善患者的预后[2,6],但是既往组织病理学分型不能很好地阐明其预后的差异性。随着分子生物学的发展,根据基因组学、蛋白组学等技术的进步,可以更好地探MB发生的分子学因素,识别潜在的分子靶点,并拟定更为精准的个体化治疗策略。分子病理在MB的诊断及预后中将占有越来越重要的地位,结合神经影像学表现可以更好地区分肿瘤的突变特征,并有助于阐明是否存在真实进展,为临床医生拟定治疗策略提供更为精确的循证医学证据。在今后的临床医学工作中,对MB分子病理进行深入研究,指导患者个体化治疗,进一步推动MB诊治的发展。
猜你喜欢
中国兽医学报(2022年9期)2022-11-11
中国生物制品学杂志(2022年3期)2022-05-13
中南药学(2022年2期)2022-03-30
中国社区医师(2019年1期)2019-06-26
上海医药(2016年6期)2016-04-28
企业导报(2015年16期)2015-12-14
智能制造(2015年4期)2015-05-12
中国青年(1990年12期)1990-08-28
