
点缺陷工程策略助力TiO2光催化分解水产氢
2022-01-27 13:39薛晋波高佳琦
太原理工大学学报 2022年1期
薛晋波,高佳琦,b
(太原理工大学 a.新材料界面科学与工程教育部重点实验室,b.材料科学与工程学院,太原 030024)
利用可再生的绿色能源取代化石燃料,是解决环境污染和能源危机的有效途径之一。众多可再生能源中,太阳能由于清洁、充足、安全等优点,被视为人类可持续发展的终极能源。考虑到太阳辐射的间歇性和扩散性,将太阳能转化为易于储存的化学燃料已成为太阳能利用的理想方案。在潜在的太阳能转化方案中,利用半导体的“人工光合作用”将水分解制取绿氢(H2)对于解决目前严峻的化石能源短缺与环境污染问题极具前景[1-2]。然而,光催化水分解是一种上坡反应,涉及多电子、多步骤反应,这对催化剂材料的设计提出了更高的要求,不仅需要其具备合适的能带结构,更需保证其光生电荷的高效分离与传输[3-4]。
光催化剂的活性与其电荷(电子和离子)传输行为及活性中心的氧化还原能力密切相关,因而利用点缺陷工程调控光催化剂的电子结构和表/界面构型,使其具有良好的光吸收能力与高效的载流子分离、传输特性[5-7]。此外,缺陷工程可以增强多组分间的界面相互作用,且多种缺陷间的协同作用可实现光催化剂的多功能化。因此,基于缺陷工程调控的光催化技术在分解水产氢领域具有广阔的应用前景[8-9]。
在现有的光催化剂体系中,借助缺陷工程提高光催化产氢效率已被广泛研究[10]。尽管如此,仍需进一步厘清缺陷工程设计中的核心问题,即如何在催化剂内精准构筑点缺陷以实现催化剂活性的提高。例如,表面缺陷可作为催化反应的活性中心以增强电子捕获,从而促进光激发电子和空穴的分离;同时,高密度的体相缺陷会作为电荷湮灭中心加剧非辐射复合和缩短载流子寿命[11-16]。另一方面,单一的点缺陷难以同时实现能带结构和高反应驱动势垒间的相互匹配,因而迫切需要将具有协同功能的多种点缺陷精确地构筑于光催化剂中,避免单一缺陷导致的掣肘效果,达到多功能缺陷协同实现催化反应中热力学与动力学动态平衡的目的。
基于此,以TiO2为研究模型,首先提出一种简易可行的点缺陷构筑策略以实现对TiO2中氧空位浓度及空间分布的调控。在此基础上,探索在单组分TiO2催化剂内构筑多元缺陷物种协同实现光催化活性增强的机理,并阐明缺陷结构与催化反应间的“构效关系”,为含缺陷催化剂的设计提供理论依据和应用指导。本文将从实验到结果讨论详细介绍这一系列研究。
1 实验
1.1 催化剂的制备
1.1.1TiO2纳米管阵列的制备
利用阳极氧化法制备TiO2纳米管阵列。用丙酮、乙醇和去离子水对钛(Ti)片进行超声清洗,然后在100 mL含NH4F的乙二醇(EG)溶液(0.74 g NH4F、98 mL EG和2 mL去离子水)中进行阳极氧化,其中Ti片为阳极,铂(Pt)片为对电极,在50 V电压下氧化1 h.阳极氧化后的样品用去离子水清洗并干燥保存,此时得到的催化剂记为未处理样品(untreatment).
1.1.2含氧空位TiO2纳米管阵列的制备
将未处理的TiO2纳米管阵列(untreatment)放置于带盖的坩埚中,使用铝粉完全包裹,然后置于真空管式炉中,在氩气气氛中600 ℃铝热还原退火2 h,得到富含氧空位的TiO2纳米管阵列,该样品标记为vo-b-TNT.
1.1.3仅含表面氧空位TiO2纳米管阵列的制备
将未处理的TiO2纳米管阵列放置于石英舟中,在空气气氛中600 ℃退火2 h,此时样品标记为TNT。随后,将TNT放置于带盖的坩埚中,使用铝粉完全包裹,在氩气气氛中600 ℃铝热还原退火2 h,得到仅含表面氧空位的TiO2纳米管阵列,该样品标记为b-TNT.
1.1.4具有氧空位/N掺杂的双缺陷TiO2纳米管阵列的制备
首先,在1.1.1步骤中的电解液内加入1.5 g尿素作为N源,并沿用上述阳极氧化工艺,制备N掺杂的TiO2纳米管阵列。随后,将其按1.1.3中的工艺进行退火处理,得到具有氧空位/N掺杂双缺陷的TiO2纳米管阵列,该样品标记为N-b-TNT.
1.2 材料表征
利用场发射扫描电子显微镜(FESEM,JEOL JSM-6700F)和高分辨率透射电子显微镜(HRTEM,JEOL JEM-2100F)表征催化剂的微观形貌和结构;采用X射线衍射仪(XRD,DX-2700X)对样品的晶体结构进行表征;通过X射线光电子能谱(XPS,Amicus Budget)分析样品的表面状态和化学组成;利用紫外-可见分光光度计(UV-vis,PerkinElmer Lambda950)分析样品的光吸收特性;利用电子顺磁共振波谱仪(EPR,JEOL JES-FA200)分析样品的缺陷状态;通过自组装表面光电压(SPV)系统对样品的电荷迁移行为进行表征;使用Autolab电化学分析仪测试样品的电化学性能。
1.3 光催化性能测试
光催化制氢反应在独立的密闭玻璃反应器中进行,采用配有紫外截止滤光片的300 W氙灯(CHF-XQ300W)作为可见光(λ≥400 nm)光源。将试样负载3% Pt(光接收面积约为2 cm2,相当于5 mg TiO2)置于100 mL甲醇/水溶液中(CH3OH/H2O体积比为1/5).光照前,用高纯度N2(99.99%)除去溶液和反应器中的残留气体,使用循环水浴将反应温度保持在室温。连续光照后,产生的H2用气体注射器取样,通过Agilent 8890型号在线气相色谱仪进行测试,采用外标法分析H2的产量与生成速率。
1.4 理论计算
本研究中的所有计算都是使用Materials Studio中CASTEP模块进行的。采用基于DFT的平面波超软赝势,通过广义梯度近似(GGA)下的PBE函数计算交换相关能量。计算的截断能为380 eV,并利用Monkhorst-Pack方式来确定布里渊区,选用3×3×1的k点网格。力收敛值为0.3 eV/nm,能量收敛值为2×10-6eV/atom.
2 结果与讨论
2.1 含氧空位TiO2纳米管阵列的表征、性能及机理探究
2.1.1结构表征
结构表征结果如图1所示。图1(a)为vo-b-TNT在Ti基底上的FE-SEM照片。TiO2纳米管在Ti基底表面均匀分布,并保持规整且高度有序的阵列结构。这种一维纳米结构一方面增大了材料的比表面积,另一方面利于载流子的定向传输。HRTEM图像显示铝热还原反应后TiO2仍具有清晰的晶格条纹,证实催化剂具有良好的结晶度,相邻晶格条纹的间距为0.352 nm,对应于锐钛矿(Anatase)(101)晶面,如图1(b)所示。通过XRD探究样品的晶体结构与物相组成(图1(c)),铝热还原反应后得到的样品高度结晶,并展现出与锐钛矿相TiO2匹配的衍射峰,这与HRTEM分析结果相一致。值得注意的是,在33.8°观测到一个与氧空位缺陷相关的衍射峰(TiO2-x),其可初步证实在vo-b-TNT中存在氧空位。

图1 vo-b-TNT样品的(a) SEM图像,(b) HRTEM图像;vo-b-TNT和untreatment样品的(c) XRD图谱,(d) 高分辨Ti 2p XPS图谱,(e) 高分辨O 1s XPS图谱,(f) EPR图谱Fig.1 (a) SEM image and (b) HRTEM image of vo-b-TNT; (c) XRD patterns; (d) Ti 2p and (e) O 1s XPS spectra and (f) EPR spectra of vo-b-TNT and untreatment sample
利用X射线光电子能谱(XPS)表征vo-b-TNT表面元素的化学状态,如图1(d)所示。对元素的高分辨图谱分峰拟合后,在结合能为457.6和463.3 eV处拟合出额外的小峰,分别对应于Ti3+2p3/2和Ti3+2p1/2[17].Ti3+的出现是由于氧空位导致Ti4+离子周围电子云密度增加,结合能降低,因此Ti3+的存在与氧空位的形成密切相关。图1(e)为vo-b-TNT和untreatment样品的O 1s高分辨图谱峰,位于结合能为529.8[18]、531.6[19]和533.1 eV[20](标记为O1、O2和O3)的三个O 1s峰可分别对应于晶格氧、缺陷氧和表面吸附氧物质。可以发现,vo-b-TNT的表面氧缺陷(O2)含量明显高于untreatment样品,表明其具有更高的氧缺陷浓度。进一步通过EPR测试,可观测到g=2.001的信号值,证实了催化剂中缺陷物种为氧空位(图1(f))[21].
2.1.2性能分析
光吸收能力是催化剂性能优劣的先决因素,分析结果如图2所示。首先借助UV-vis DRS分析了vo-b-TNT和untreatment样品的光响应能力(图2(a)).由于锐钛矿型TiO2固有的宽带隙(Eg=3.2 eV),使得untreatment样品只能吸收紫外光。与untreatment样品相比,vo-b-TNT表现出强烈的全光谱吸收,其吸收范围扩展到可见甚至红外光区。这种显著增强的光吸收能力归因于大量存在的自掺杂氧空位形成缺陷能级,减小了TiO2的带隙。
载流子的分离与传输效率是影响材料光催化性能的另一重要因素。为了探究含氧空位TiO2中光生载流子的行为,进行了瞬态光电流测试(图2(b)).通过缺陷调控的vo-b-TNT在可见光照射下的光电流明显高于untreatment样品。较高的光电流强度表明vo-b-TNT中高效的电荷分离和转移。然而值得注意,vo-b-TNT具有高的暗电流值,究其原因,可认为vo-b-TNT存在高浓度的氧空位而形成重掺杂状态,这种重掺杂态使vo-b-TNT发生简并现象而表现出类金属特征。总而言之,引入氧空位后的vo-b-TNT光电流强度较未改性前显著提升,这种光电性能的改善一方面归因于vo-b-TNT对可见光吸收的增强,另一方面主要是氧空位提供活性位点促进了电荷分离。
为了评价vo-b-TNT的光催化性能,研究了样品在可见光下的光催化产氢活性。如图2(c)所示,在5 h的持续光照下,vo-b-TNT表现出明显增强的产氢活性,其平均产氢速率为657.35 μmol·g-1·h-1,比untreatment样品提升8倍以上。产氢性能的提升一方面是由于vo-b-TNT具有宽的光响应范围,使得光生载流子浓度增大;另一方面,氧空位易于捕获光生电子,从而促进电荷的分离与转移。

图2 未处理样品和vo-b-TNT的(a)UV-vis DRS光谱,(b)光电流图谱及(c)光催化制氢性能Fig.2 (a) UV-vis DRS spectra, (b) photocurrent density, and (c) hydrogen evolution performance of untreatment sample and vo-b-TNT
2.1.3机理探究
借助DFT理论计算模拟可以辅助探究反应机理,结果如图3所示。本文构建了有/无氧空位两种锐钛矿型TiO2模型,并以此进行了H2O分子吸附构型及解离反应路径的探索,如图3(a)所示。H2O分子在无氧空位锐钛矿型TiO2(101)表面的最稳定吸附构型是与一个Ti5c原子结合,并与两个相邻的O2c原子形成弱氢键。在这种状态下,吸附能为0.693 eV.最稳定的解离构型是H2O分子的一个氢原子与相邻的O2c原子形成羟基(—OH)键,其余的—OH通过氧原子与表面Ti5c原子键合。在这种状态下,吸附能为0.400 eV,表明该过程是吸热反应。反应过渡态的吸附能为0.026 eV.从最初的吸附分子状态,H2O分子解离需要0.719 eV的总活化能,而其逆反应的活化能为0.426 eV.因此,H2O分子在完美锐钛矿型TiO2(101)表面解离更困难。当一个H2O分子吸附在单层氧空位浓度为1/8 ML的锐钛矿TiO2(101)表面的相同位置时,如图3(b)所示。通过优化吸附模型获得最稳定的吸附构型。H2O分子的O原子几乎占据了空位桥接O2c原子的位置,一个氢原子指向相邻的桥接O2c原子,另一个指向远离表面。通过分析H2O分子在不同表面解离的过渡态,发现在氧空位浓度为1/8 ML的表面上,一个H2O分子需要吸收1.183 eV的能量才能达到过渡态,而逆反应活化能为1.837 eV.在含缺陷的表面发生水分解的逆反应的活化能远大于正反应的活化能,因此反应总体上是放热的,即引入氧空位后,反应更倾向朝解离方向进行,因此氧空位可视为分解H2O分子的活性位点。
基于上述分析与讨论,提出了具有单一氧空位自掺杂改性TiO2的光催化分解水产氢机理。如图3(c)所示,通过在缺氧条件下进行铝热还原处理,高温加速了晶格振动,并促使Ti-O键破坏,Al驱动TiO2失去晶格氧而产生氧空位。首先,这些氧空位可在带隙中引入缺陷能级,降低电子跃迁所需能量,促进TiO2对可见光的吸收;其次,氧空位作为浅电子陷阱,避免了载流子的复合,同时显著增加了载流子浓度,提高了光生电荷的分离、传输与利用效率;最后,借助理论计算证实氧空位可作为表面反应的活性中心,促进H2O分子的解离以形成H2.综上所述,氧空位的引入促成TiO2光催化要素获得全面改善,因此构筑氧空位可视为一种有效的改性策略。

图3 H2O在氧空位浓度为0(a)和1/8 ML(b)的锐钛矿型TiO2 (101)表面的吸附构型和分解反应途径;(c)含氧空位的TiO2的光催化制氢机理Fig.3 Adsorption configurations and decomposition reaction pathway of water on anatase TiO2 (101) surface with 0 (a) and 1/8 ML OVs (b); and (c) mechanism of photocatalytic hydrogen evolution over TiO2 with oxygen vacancy
2.2 含表面氧空位与晶格应变的TiO2纳米管阵列的表征、性能及机理探究
尽管直接利用铝热还原法在TiO2中引入的氧空位在一定程度上促进了材料的光吸收、电荷分离与表面反应,然而未经过精准调控的氧空位可能由于其不当的掺杂浓度及空间分布使得在TiO2晶体内部引入重掺杂的体相氧空位,而体相氧空位常被视为电荷的复合中心抑制载流子在催化剂体相中的分离与传输[11]。因此有必要改进制备TiO2的工艺,合理调控氧空位的空间分布,进一步促进TiO2纳米管阵列中光生电荷的高效转移。
2.2.1结构表征
b-TNT和vo-b-TNT的结构表征结果如图4所示。为了探明TiO2内缺陷的存在形式与分布规律,厘清缺陷所处的化学环境及其附近原子的键合结构,采用XPS深度剖面技术对氩气刻蚀前后的样品进行电子结构分析,用来表征材料表面和体相的化学环境和电子结构。如图4(a)所示,vo-b-TNT与b-TNT中Ti 2p在位于463.7 eV和458.0 eV的谱峰来自Ti4+2p1/2和Ti4+2p3/2自旋轨道分裂产生的光电子[22];此外,位于462.7 eV和457.0 eV的谱峰分别对应于Ti3+2p1/2和Ti3+2p3/2[22].图4(b)为经Ar气刻蚀后催化剂体相元素的化学键合结构。与表面XPS分析结果对比,仅在vo-b-TNT中观察到了与缺陷存在有关的Ti3+峰,在氩气刻蚀后b-TNT的体相Ti 2p XPS谱中未观察到与氧空位相关的Ti3+存在。XPS峰面积可代表被测元素的相对含量,对刻蚀前后样品的XPS谱图进行对比发现:b-TNT仅在表面形成了氧空位,并无体相氧空位存在。
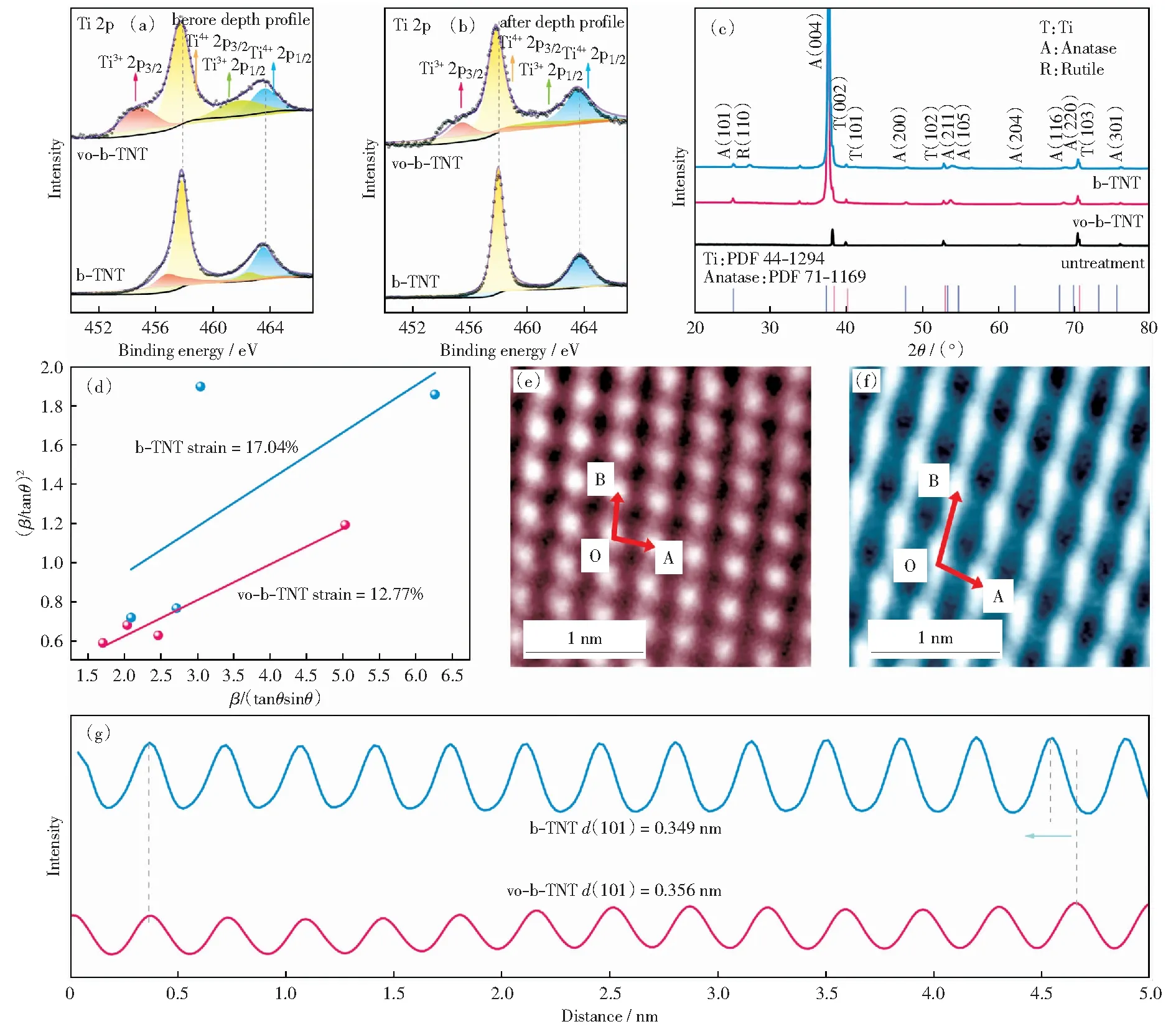
图4 b-TNT 和vo-b-TNT深度剖面(a)前与(b)后的高分辨Ti 2p XPS光谱;上述两种催化剂的(c)XRD图谱;(d)Halder-Wagner曲线;(e,f)HRTEM图像;及(g)随TiO2(101)空间方向的综合像素强度[23]Fig.4 High-resolution XPS spectra of Ti 2p before (a) and after (b) depth profile; (c) XRD patterns; (d) Halder-Wagner plot; (e,f) HRTEM images and (g) integrated pixel intensities of b-TNT and vo-b-TNT[23]
利用X射线衍射(XRD)谱进一步厘清表面氧空位对催化剂晶体结构和相组成的影响。vo-b-TNT与b-TNT的XRD图谱显示出相近的晶体结构,其均以锐钛矿相为主,这表明不同的氧空位分布不会改变TiO2纳米管的晶体结构(图4(c)).利用Bragg定律得到了不同样品的晶格参数如表1所示。根据计算结果可知,b-TNT晶体可能由于“外延”失去晶格氧,“强制”膨胀的c轴将诱导产生拉伸应变,同时晶格参数a和b减小以保持其单胞体积,晶格参数的明显变化证实应变的存在。

表1 两种光催化剂的晶格参数Table 1 Lattice parameter of two types of photocatalyst
构建表面应变是调节金属氧化物催化剂反应活性的有效策略。根据XRD结果可以计算出材料的应变程度。进一步采用Halder-Wagner公式对催化剂内部存在的应变进行计算[24],如公式(1)所示:
(1)
其中:εHW和DHW分别表示TiO2纳米管阵列薄膜的应变和晶粒尺寸;K=0.9为形状因子;β、λ、θ分别为半宽峰、Cu Kα辐射波长(1.540 6 nm)与X射线衍射角。
如图4(d)所示,晶格应变为正,表明晶格受到拉伸力的影响。从计算结果和H-W图可以发现,与vo-b-TNT相比,b-TNT表现出明显的晶格应变,这归因于在空气气氛中预退火后已结晶的TiO2其晶格氧在持续的铝热还原处理下被“强制”移除而形成表面氧缺陷。此外,完全结晶的亚表面与有缺陷的表面结合,在界面处不可避免地形成晶格失配,从而在b-TNT中形成显著的残余拉伸应变。
作用在晶体上的内应变会影响晶格原子的排列,由于应变的存在,原子间距离会拉长/缩小,从HR-TEM图像可知(图4(e),(f)),vo-b-TNT和b-TNT的晶格参数不仅表现在晶格向量的长度不同,且晶格向量之间的夹角也存在差异。此外,图4(g)显示了沿TiO2(101)方向的13层原子的积分像素强度,计算出vo-b-TNT和b-TNT的平均晶面间距分别为0.356 nm和0.349 nm.由此定量分析了由氧空位引起的晶格应变,也证实了在表面氧缺陷诱导下形成的剪切应力致使晶格矢量发生改变。
2.2.2性能分析
通过UV-vis DRS分析了氧空位的分布对催化剂光吸收能力的影响如图5所示。从图5(a)可以看出,所有样品在紫外区均表现出强烈的响应信号。此外,由于大量氧空位的存在,在TiO2导带下方引入了一个新的缺陷能级,降低电子跃迁所需的能量,因此,含氧空位的TiO2在250 nm到800 nm的波长范围以及在红外区域表现出连续的光吸收,由此可知,表面和体相氧空位均有助于改善TiO2的光捕获能力。
利用表面光伏技术(SPV)分析了样品表面光生电荷转移的行为,图5(b)中所示的正向光伏信号符合n型半导体的特性[25]。光激发的正电荷(h+)向表面迁移,导致材料表面和体相之间产生正电位差。无缺陷的TiO2纳米管阵列(TNT)显示出最高的光电压值,表明在紫外光区域,TiO2具有高的载流子生成及分离效率。随着氧空位的引入,TiO2的光电压值明显降低,这归因于氧空位的存在改变了电荷传输行为,更多电子向表面迁移导致正信号被抑制。值得注意的是,存在高浓度氧空位的vo-b-TNT的光电压信号甚至无法被检测到,究其原因,高密度的体缺陷可显著降低光催化剂的内阻而促进载流子的传输,但其在一定程度上抑制了光生电荷在光催化剂表面的堆积,不利于表面反应物的活化。作为对比,具有表面氧空位与应变效应协同调制的b-TNT则实现了光生电荷传输与表面反应间的动态平衡。
在可见光照射下进行光催化水分解测试如图5(c)所示,vo-b-TNT和TNT的光催化产氢平均速率分别为660和147 μmol·g-1·h-1,在表面氧空位和晶格应变的双重调制下,b-TNT获得了高达1 882 μmol·g-1·h-1的优异产氢速率。这种显著增强的产氢效率进一步证实了表面氧空位自掺杂结构和内部晶格应变之间良好的协同作用。
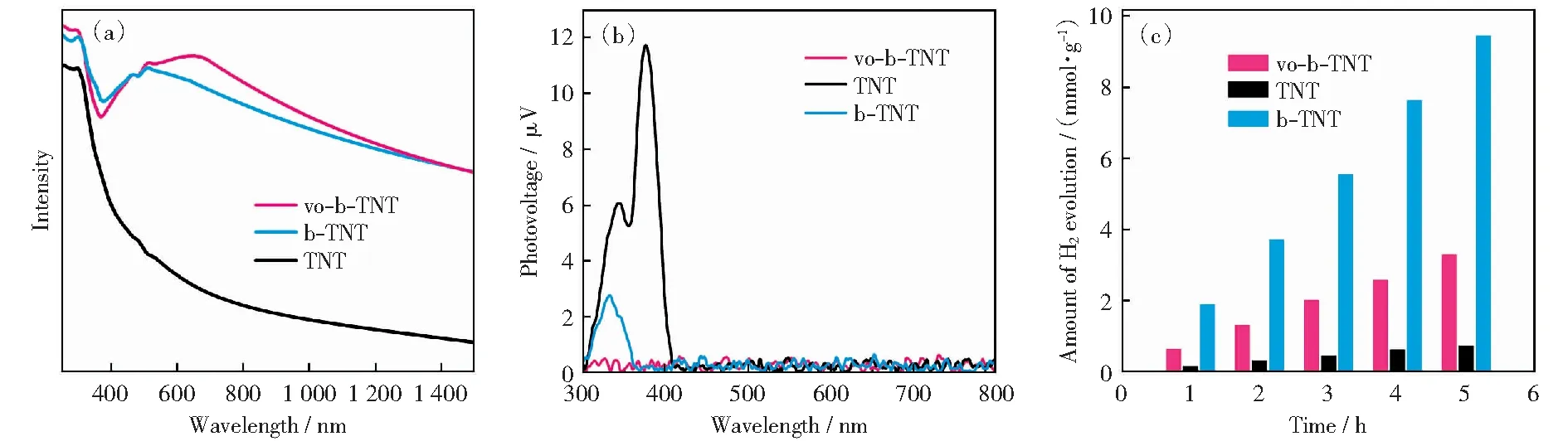
图5 试样的(a)紫外-可见漫反射吸收光谱,(b)SPV图谱及(c)光催化制氢性能Fig.5 (a) UV-visible DRS spectra, (b) SPV spectra, and (c) hydrogen evolution performance of the samples
2.2.3机理探究
基于密度泛函理论(DFT)的仿真计算是预测材料结构与性能之间内在联系的有力工具。考虑氧空位和晶格应变的存在,构建并优化了锐钛矿、具有氧空位的锐钛矿和具有表面氧空位与应变协同作用的锐钛矿三种晶体模型如图6所示。图6(a)-(c)为三种晶体模型的2D差分电子密度(EDD)横截面图。氧空位的存在改变了材料局部电荷分布,使原本富集在氧原子处的电子向其附近发生转移。应变的存在会加强这种电荷转移过程,促使氧空位邻近的Ti原子周围富集更多的转移电荷。基于EDD分析,通过设计氧空位和应变共存的结构并耦合二者优势,可协同实现TiO2中电荷转移过程的优化,并进一步促进光催化产氢。
根据上述实验和计算结果,提出b-TNT光催化性能增强的机制,如图6(d)所示。一方面,氧空位的引入可有效地缩小带隙,提高b-TNT的可见光利用率。在b-TNT光催化剂体系中由于缺氧,更多的键合电子转化为自由电子,而表面氧空位带正电,捕获并富集受激发的电子和自由电子。此外,由于配位不饱和的表面氧空位可作为分子吸附和反应的活性位点,降低b-TNT光催化产氢的势垒。另一方面,TiO2中完美的体相晶格(图6(d),区域1)与含有氧空位的表面晶格结合,导致界面处的晶格失配,促使应变的发生,其表现为Ti-O键的扭曲和晶格常数的变化(图6(d),区域2).由表面氧空位诱导产生的晶格应变促使高能表面结构形成,其为电子的定向传输提供了驱动力和通道,并促使电子向催化剂表面迁移。由此可见,通过氧空位和晶格应变的协同效应促进电荷的产生、分离和迁移,从而最终增强b-TNT光催化析氢性能。
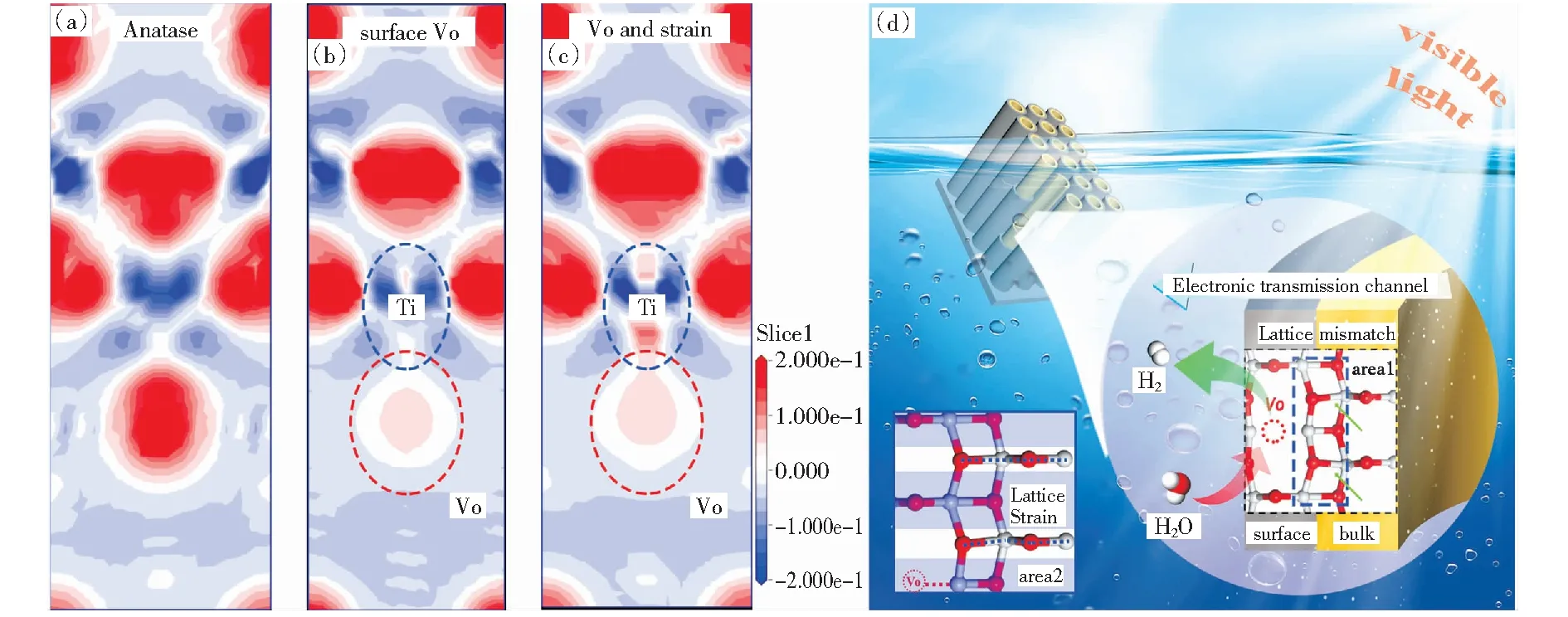
图6 (a-c)锐钛矿三种晶体结构模型的2D差分电荷密度;(d)可见光照射下b-TNT体系的电荷转移和光催化过程[23]Fig.6 (a-c) 2D charge density difference cross-section of the Vo-section; and (d) proposed mechanism for charge transfer and photocatalytic process in b-TNT system under visible light irradiation[23]
2.3 含表面氧空位/体相N掺杂双缺陷TiO2纳米管阵列的表征、性能及机理探究
TiO2中合理地引入表面氧空位在一定程度上改善了电荷分离与传输特性,然而由于表面氧空位诱导的应变效应存在局部性与随机性,使得光生电荷转移的驱动力有待提升。此外,表面氧空位对催化剂能带结构的调控也有限。因此,在单组分TiO2中合理地构筑具有协同功能的多元缺陷结构,可实现光捕获的增强且避免制氢过程中热力学势垒的损失;同时引入晶体内建电场,加速TiO2中光生电荷的定向转移。
2.3.1结构表征
双缺陷对TiO2晶体结构的影响的表征如图7,对不同样品进行了XRD分析,如图7(a)所示。催化剂均以锐钛矿型TiO2为主,随着N的掺杂,锐钛矿(101)晶面的衍射峰强度降低,这可能是由于N的掺杂和其诱导产生的氧空位影响了晶体结构的周期性。借助电子顺磁共振(EPR)技术进一步探测由缺陷引起的未成对电子,以确定缺陷物种。如图7(b)所示,在样品中观察到了g因子为2.001的未成对电子,对应于氧空位物种[21],且EPR信号强度随着N掺杂引入而增强,即晶体中N的引入将进一步增加氧空位的浓度。
由于光催化反应与材料表面的化学成分和状态密切相关,利用X射线光电子能谱(XPS)分析了氧空位和N掺杂对材料表面的化学成分和相应的电子结构的影响。如图7(c)所示,高分辨率Ti 2p图谱中Ti 2p的宽峰对应于Ti4+与Ti3+两对峰[22]。利用Ar气深度刻蚀后仅观测到Ti4+的反褶积峰(图7(d)),证实制备的催化剂仅存表面缺陷(Ti3+/Vo).此外,与b-TNT样品相比,N-b-TNT的结合能位置向更低的能量偏移,且Ti3+的反褶积峰面积明显增大。一方面由于在O-Ti-N结构中,电子从N原子转移至Ti原子,Ti的电子云密度增大从而使其结合能降低;另一方面,Ti3+的反褶积峰面积与氧缺陷的浓度相关,Ti3+峰面积的变化表明为了平衡N掺杂带来的电荷失衡,更多电子转移至表面,表现出更强的Ti3+信号。由高分辨率O 1s谱图可知,氧元素对应于晶格氧、缺陷氧、表面吸附羟基氧[22]的三个峰(图7(e)).与b-TNT相比,N-b-TNT中观测到N掺杂相关的反褶积峰存在(图7(f));此外,缺陷氧在体相的反褶积峰值弱于其在表面的强度,这证实了N-b-TNT具有独特表面氧空位与体相N掺杂的结构。此外,催化剂表面均未检测到N的存在(图7(g)),而在体相N 1s图谱中395~403 eV范围检测到N信号(图7(h)),结合能位于399.2和399.9 eV处分别对应于取代型Nsub(O-Ti-N)和间隙型Nint(Ti-O-N)[26].Nsub位于低结合能位置意味其具有更高电子云密度,该氮原子处于负氧化状态且其电荷实际上减少到约为-1[27],基于此可推断出取代型Nsub通常可作为电子供体为光催化反应提供电子。XPS深度剖面结果证实了N-b-TNT存在表面氧空位与体相Nsub掺杂两种层级分布双缺陷,二者将协同耦合进一步影响催化剂电荷分离与转移行为。
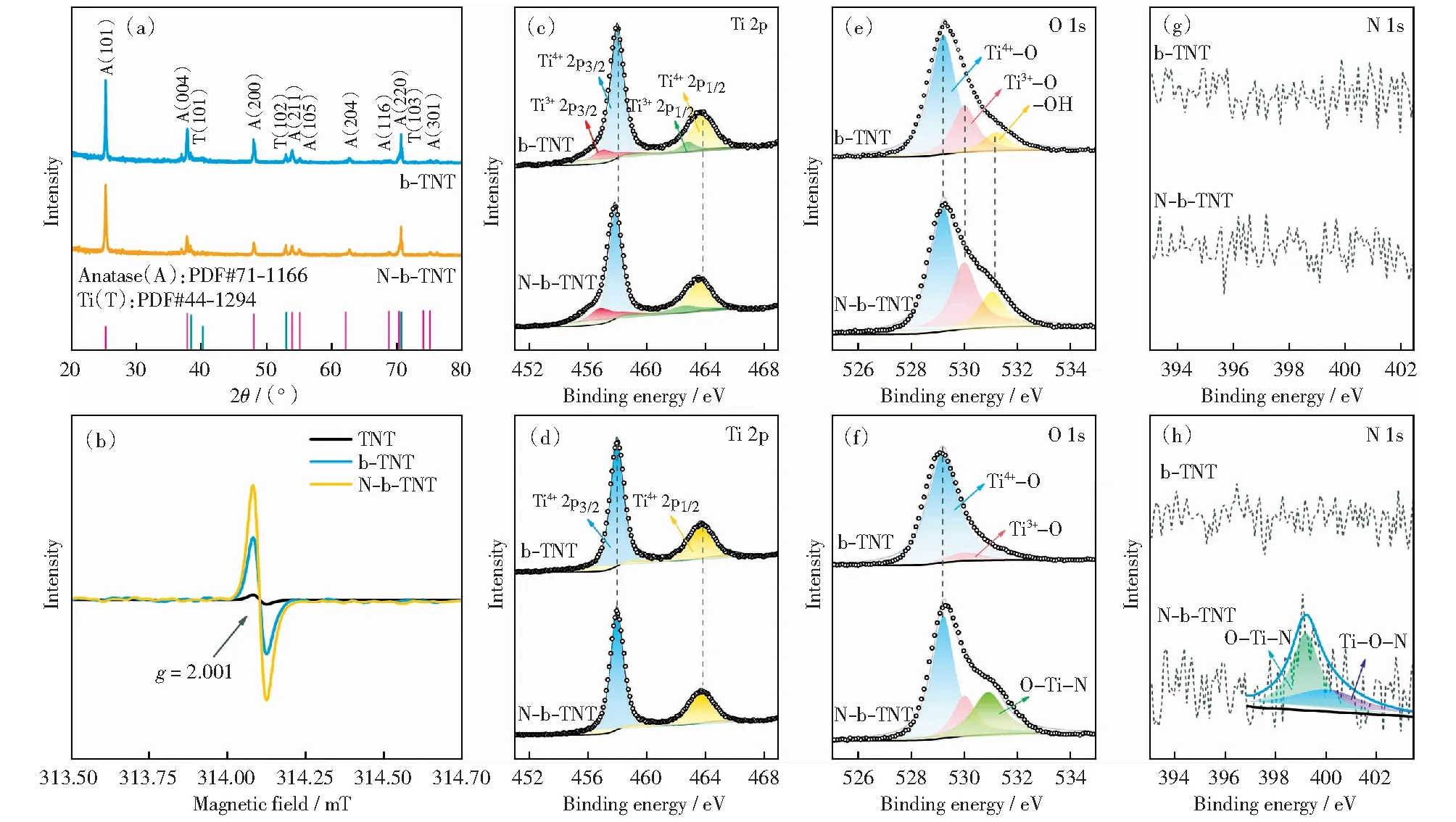
图7 含双缺陷的TiO2的(a)XRD图谱与(b)EPR图谱,深度剥层前(c)Ti 2p(e)O 1s和(g)N 1s的高分辨XPS光谱,深度剥层后(d)Ti 2p(f)O 1s和(h)N 1s的高分辨XPS光谱[28]Fig.7 (a) XRD patterns and (b) EPR spectra at room temperature; high-resolution XPS spectra of Ti 2p before (c) and after depth profile (d), O 1s before (e) and after depth profile (f), and N 1s before (g) and after depth profile (h), corresponding to the surface and bulk phase analysis, respectively[28]
2.3.2性能分析
N-b-TNT的光催化性能分析结果如图8所示。利用UV-vis DRS证实,经过双缺陷调制后的TiO2光吸收范围得到了明显的拓宽,如图8(a)所示。尽管光吸收能力的提升是增强光催化性能的前提,但受激电荷仍需要具备合适的电位才能参与光催化反应。从热力学角度考虑,光催化产氢反应要求光催化剂导带电势较H+/H2电势(0 V vs NHE)更负,且更负的导带位置意味着光生电子具有更强的还原能力,这对合理地设计光催化剂提出了要求:催化剂在带隙宽度与氧化还原动力间需要权衡。基于此,考察了N-b-TNT的导价带位置。由表面VB-XPS图谱结果确定了催化剂的价带(VB)位置,如图8(b)所示。显而易见,N掺杂是价带顶抬高的主要因素,N 2p和O 2p重叠导致VB上方杂化轨道出现,其促进催化剂带隙缩减并增强对可见光的捕获。根据Mott-Schottky线性电势曲线的截距值(图8(c)),判定b-TNT和N-b-TNT的平带电位分别位于-0.28与-0.30 V(vs NHE,pH=0).一般来说,n型半导体的导带(CB)位置接近于Mott-Schottky测试所得平带电位[29]。因此,可以明确的是,催化剂的导带位置得到了保持,被激发的电子仍具有足够的电位势垒,可以用来驱动光催化产氢反应。双缺陷调控的N-b-TNT实现了光捕获的增强,且避免了产氢过程中热力学势垒的损失。

图8 含双缺陷TiO2的(a)紫外-可见漫反射吸收光谱,(b)表面VB XPS图谱,(c)Mott-Schottky图及材料的平带电位,(d)SPV光谱及其相位角,(e)SPV信号矢量相加示意图及(f)光催化制氢性能[28]Fig.8 (a) UV-visible diffuse reflectance spectroscopy; (b) valence and (c) conduction potential; (d) SPV spectrum and its phase; (e) SPV signal additivity sketch map; and (f) hydrogen evolution performance of TiO2 prepared by dual-defect regulation[28]
为了进一步研究载流子转移动力学,进行了表面光电压(SPV)和其相位角分析。随着N掺杂量与氧空位浓度的增加,双缺陷调制的N-b-TNT样品中SPV信号强度整体降低(图8(d)).简单来讲,正的SPV信号意味着正电荷在表面的富集,导致界面处正电势差的存在,降低的正信号可归因于电子向表面的转移与注入。利用相位角可以更好地分析电荷的转移过程。通常,随着电子(空穴)向催化剂表面移动的趋势增加,相位角逐渐向180°增加(向0°减小)[30-31]。SPV结果表明,N-b-TNT样品的SPV值最低且具有更正的相位角。作为典型的n型半导体,TiO2材料的SPV相位理论上应在-90°~0°之间,而N-b-TNT样品的相位在第一象限,这表明第二象限中必须存在另一个属于相反电场的信号且相位角趋向180°(参见图8(e)),该电场由表面氧空位与体相掺杂的N协同诱导产生,且电场方向为由表面氧空位指向体相N,其与电子运动方向相反,该结构可促进光生电子向表面迁移。
由光催化产氢性能测试可知,具有双缺陷调控的N-b-TNT样品其产氢速率达到3 183 μmol·g-1·h-1,约是b-TNT的1.8倍和原始TNT的21倍。为了验证N-b-TNT样品的光催化稳定性,对其进行了五次光催化产氢实验循环,如图8(f)所示。由图可知,第5次氢析出速率与第一次相比衰减甚微,说明N-b-TNT具有良好的光稳定性。
2.3.3机理探究
N-b-TNT的光催化性能增强的机理分析结果如图9所示。图9(a)为含表面氧空位(Vo)及体相掺杂N的N-b-TNT模型的原子态密度(DOS)图。随着N原子与氧空位的掺入,在双缺陷调制的N-b-TNT模型DOS中观察到带隙缩减至2.21 eV,除在导带下方具有与Vo的引入相关的杂质能级外,由于O 2p与N 2p轨道杂化引发价带顶提升,这是N-b-TNT可见光吸收显著增强的主要原因。为了验证N和Ti-O之间存在的相互作用,进一步分析了N-b-TNT的分波态密度(PDOS),如图9(b)所示。N 2p的能量与O 2p的能量匹配较好,N 2p态与O 2p态的充分重叠表明了存在p-p轨道杂交,这促进N杂质态的离域化,N可作为电子供体使其在电荷寿命内被激发,并进一步转移至催化剂表面的反应位点参与产氢反应[32]。
当体系存在Vo时,与Vo相邻的低配位Ti原子(Ti5c)可视为结构模型的活性位点来接受Vo留下的多余电子。由于电子转移,TiO2(101)表面对吸附物的结合与活化能力会增强,从而促进产氢反应进行。为了可视化Vo和N掺杂对TiO2(101)表面电荷转移的贡献,对N-b-TNT模型的电荷差分密度进行了计算以厘清电子迁移规律。与仅含氧空位的b-TNT相比,N-b-TNT的Ti5c表现出明显的电子富集,如图9(c)、(d)中区域Ⅰ和Ⅱ所示。此外,对比区域Ⅲ和Ⅳ处的电子云密度,证实了体相N掺杂是更富电子的,这些电子在内建电场的驱动下,从具有高电子密度的体相向TiO2(101)面的活性位点Ti5c迁移。
综上所述,N-b-TNT优异的光催化活性与双缺陷的存在及其层级分布的独特结构相关,如图9(e)所示。双缺陷通过p轨道调制在提高催化剂光捕获率的同时,维持了导带电位,确保了被激发电子具有足够的热力学反应势垒驱动光催化产氢反应。更重要的是,该结构规避了单缺陷可能成为电荷复合中心的弊端。具有层级分布的双缺陷在表面和体相之间由于带电性质的差异,形成内建电场,优化了载流子传输动力学并促进电荷重新排布,驱使电子向表面活性中心定向迁移,最终提高其光催化产氢活性。

图9 N-b-TNT的(a)能带结构和(b)分波态密度(PODS);(c)b-TNT和(d)N-b-TNT的侧视EDD图;(e)可见光照射下具有双缺陷调控的TiO2体系中电荷产生、转移和光催化过程的增强机理[28]Fig.9 (a) Band structure and (b) partial density of states (PDOS) for N-b-TNT; side-view electron density difference map of (c) b-TNT and (d) N-b-TNT and (e) proposed mechanism for charge generation, transfer, and photocatalytic process in dual-defect controlled TiO2 system under visible light irradiation[28]
3 结论
本研究利用铝热还原法高效地在TiO2内引入氧空位缺陷,通过调整其空间分布并耦合多元缺陷,实现光催化产氢效率的稳步提升。具体为:
1) 氧空位的引入形成中间缺陷能级降低了电子的最大跃迁能,拓宽了TiO2对可见光的吸收;另一方面,这些氧空位可作为催化反应的活性位点,在一定程度上加速电荷的转移,并促进H2O分子在催化剂表面的吸附与解离。
2) 通过对TiO2中氧空位空间分布的精确调控,获得仅含表面氧空位的TiO2,抑制了非辐射复合并延长了光生载流子寿命。与此同时,含缺陷的表面结构与完整结晶的体相结构间存在晶格失配,诱导产生内建电场,改善电荷转移行为,进一步促进催化活性的提高。
3) 精确设计了含表面氧空位/体相N掺杂双缺陷结构的TiO2,协调共存的双缺陷通过引入缺陷能级及p轨道调制进一步缩减催化剂的带隙,增强太阳光的利用;同时恒定的导带位置确保了被激发电子具有足够的热力学反应势垒驱动光催化产氢反应。此外,层级分布的双缺陷诱导催化剂体内形成更加显著的内建电场,为电子传输提供了强大的驱动力,从而获得最佳的光催化析氢性能。
猜你喜欢
光子学报(2022年6期)2022-07-27
当代作家(2021年11期)2021-12-17
疯狂英语·新阅版(2021年9期)2021-10-30
中小企业管理与科技·上旬刊(2021年6期)2021-07-14
校园英语·月末(2019年11期)2019-09-10
中国科技纵横(2019年3期)2019-03-25
科教导刊·电子版(2017年35期)2018-01-27
分析化学(2017年12期)2017-12-25
分析化学(2015年3期)2015-04-20
读者欣赏(2014年6期)2014-07-03
