
CuSnZr三元催化剂应用于NTP强化催化脱硫过程的特性
2022-01-21 02:12周正华蒋连爽张震宇宁静远宁致远
中国环境科学 2022年1期
周正华,蒋连爽,张震宇,黄 锐,宁静远,宁致远*
CuSnZr三元催化剂应用于NTP强化催化脱硫过程的特性
周正华1,蒋连爽2,张震宇2,黄 锐2,宁静远3,宁致远2*
(1.云南大学材料与能源学院,云南 昆明 650000;2.云南大学化学科学与工程学院,云南 昆明 650000;3.昆明中科云环保有限责任公司,云南 昆明 650000)
为高效地在低温下处理大量低浓度电解铝烟气,采用浸渍法制备了Sn Zr型金属氧化物并添加Cu作为助剂的催化剂,并首次测试了它在低温等离子体(NTP)技术上的脱硫效果,结果表明负载了20wt% Cu老化温度为40℃的催化剂表现出最佳的脱硫性能.并对强化之后的催化剂进行了表征,与新鲜的催化剂对比,X射线衍射分析(XRD)结果表明放电对催化剂晶型基本不产生影响;扫描电子显微镜(SEM),氮吸附和脱吸(BET)表明放电会使催化剂的吸附脱附能力与孔道结构有较大提升;X射线光电子光谱(XPS)也表明放电会使催化剂表面元素价态变化,从而使其氧化还原性能改变,反应路径发生偏向;催化剂性能理论计算表明铜含量的上升会导致催化剂能带结构改变,更好利用于激发气体.
催化剂;等离子体强化催化;电解铝烟气脱硫;DBD放电;制备;数值分析
当烟气中二氧化硫(SO2)的浓度低于2%(体积分数)时,就称之为低浓度SO2烟气.在典型工业场景中,其待处理浓度多为0.1%~0.5%.相对于成熟的高浓度SO2烟气治理方法,低浓度SO2烟气目前尚无经济可行的治理技术.如何低成本、高效的处理该烟气是一个世界性难题.因此造成了目前工业烟气中的低浓度SO2基本未净化,直接排放的现状[1].以占全云南省有色金属冶炼烟气排放量的70%的电解铝行业为例,在2019年就有41~47万t SO2未经净化直接排放,这不仅浪费了硫磺资源,也造成了严重的环境污染[2].
相较于去除高浓度SO2的湿法、半干法,低浓度SO2目前还没有经济有效的治理技术.研究发现烟气中的CO可以直接将SO2还原为硫[3-4].Han等[5-6]认为,利用CO还原SO2过程涉及氧化还原反应机制与羰基硫(COS)中间体机制两种催化反应机制.
将CO直接用来还原SO2的优点在于将污染物作为反应物,在去除污染物CO和SO2的同时还能产生有价值的单质硫.因此利用烟气中的CO作为还原剂除去烟气中低浓度的SO2是具有工业应用潜力的.但是,目前上述反应通常是采用催化的方式进行,且催化反应只有在较高温度(>400℃)才有高的转化效率[7-12],这就意味着需要对烟气进行二次加热,提高成本.催化剂高温极易烧结失活,大大缩短使用寿命.因此,寻找低温催化转化方法成为工业脱硫的一大难题.
有研究人员提出了利用外场辅助强化的方法,即利用热能以外的能量形式来充分激发反应物,使其能够在低温下进行催化反应[11].低温等离子体(NTP)强化催化技术是目前较为突出的方法,现已成功地应用于工业烟气的清洁,主要表现为介质阻挡放电[13-15].该方法的主要优点是广泛的适用性,例如在去除挥发性有机化合物(VOCs)[16-17],控制颗粒物排放[18-21]和通过液相排放对柴油进行脱硫[22-23]等方面.而等离子体辅助增强催化反应是通过刺激分子来产生振动,从而提供足够的能量来控制反应速率[24].对于催化剂,Park等[7-8]研究表明SnZr基催化剂由于脱硫时不生成COS,是进行氧化还原工业化脱硫的理想催化剂.但是SnZr基催化剂也会随着晶格氧空位的氧化而失去活性中心,同时,证实了向催化剂中添加铜减少了氧化还原反应中活性位点的需要,并提高了反应效率.
因此,本文结合催化法和等离子体法的优点,采用低温等离子体强化催化反应的方式,高效地在低温下处理大量的低浓度电解铝烟气.在本研究中,以目前报道过有较好的催化脱硫活性的Sn-Zr型金属氧化物并添加Cu作为助剂为模板催化剂,并在此基础上使用NTP技术来强化烟气脱硫.
1 材料与方法
1.1 材料
本研究使用了SnZr型金属氧化物并添加Cu作为助剂的模板催化剂来进行脱硫.首先考察了在催化剂中铜的质量分数(5%、10%、20%)和制备温度(40, 60, 80℃)对催化效率的影响.然后利用介质阻挡放电(DBD)反应器对烟气进行激发,进而研究脱硫效率.模板催化剂的组成如表1所示.
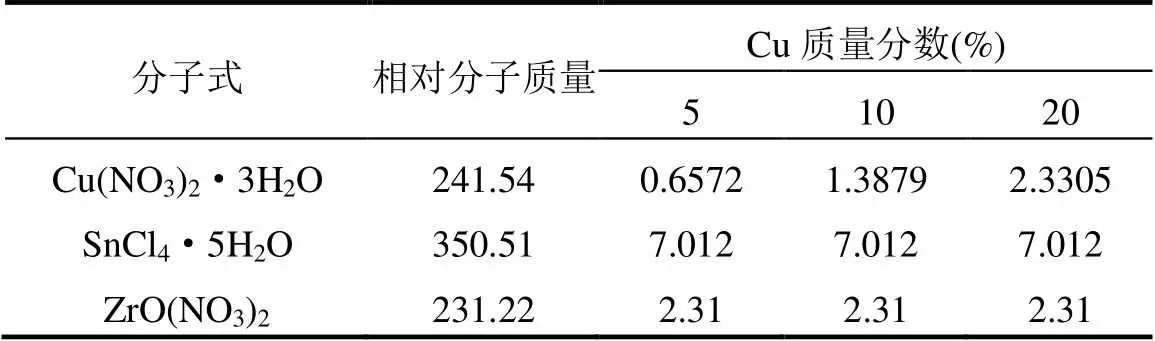
表1 催化剂组成(g)
模板催化剂采用浸渍法制备.以含铜为10%的催化剂制备为例:首先称取7.012g SnCl4、2.31g ZrO(NO3)2、1.3879g Cu(NO3)2×3H2O溶于100mL蒸馏水中,然后用氨水滴定调节溶液pH值至7,再用多孔保鲜袋封口,随后在不同的老化温度(40, 60, 80℃)下在通风橱中恒温搅拌6h.抽滤老化后得到的溶液,将滤饼置于105℃鼓风烘箱内12h烘干水分,取出后于马弗炉中以2℃/min的速度程序升温至600℃,恒温焙烧4h.对焙烧后的催化剂进行研磨、压片并筛分出30~50目均匀的催化剂颗粒留待备用.实验中所用4种化学试剂均为分析纯.
1.2 实验装置
采用室内搭建的等离子体-催化集成评价系统,研究了催化剂在DBD放电强化下对电解铝烟气脱硫效率的影响.如图1所示,集成系统由4部分组成:1)烟气模拟系统;2)DBD等离子体放电反应器系统;3)催化剂活性评价系统;4)尾气检测子系统.
如图2所示,DBD等离子体放电反应器系统: DBD反应器的外壁是一个400mm长的透明石英管,其相对介电常数为3.7,外径为25mm,内径为22mm.高压电极是直径为12mm的实心铝棒,直径为0.5mm的5m长的接地铜线缠绕在石英管的外侧壁上.DBD反应器使用南京苏曼等离子技术有限公司提供的CTP-2000K AC高压高频电源供电.DBD反应器的电特性参数收集和记录使用示波器(Tektronix, TDS2024C)和高压探头(Tektronix P6015A).如图2c所示,探针连接到两个测量位置.高压探头直接连接到电源输出端子,低压端子通过测量电容器(=0.47mF)连接到DBD反应器的接地电极.使用这些探针获取的两个信号用于生成DBD反应器的李萨如(Lissajous)图形,以计算DBD反应器的关键参数值,即功率(),气隙等效电容(g)和电介质等效电容(d).
催化剂活性评价系统:将1g Cu-SnZr催化剂放入外径为10mm,壁厚为1.5mm的石英管中.利用石棉固定催化剂,将石英管垂直放置在具有220V和1.5kW电源(SK2-1;武汉亚华电炉有限公司)的管式炉中.使用可编程控制器(带有单相智能PID触发器的YHA-5000)控制管式炉的温度,以保持催化脱硫过程所需的温度.系统还用于预硫化催化剂并评估预硫化后的催化性能.
1~6气瓶,7~12质量流量计,13单向阀,14流量计控制器,15石英管反应器,16立式管式炉,17微型电除尘器,18电除尘器电源,19 DBD反应器,20交流电源,21变压器,22保温套管,23冷凝装置
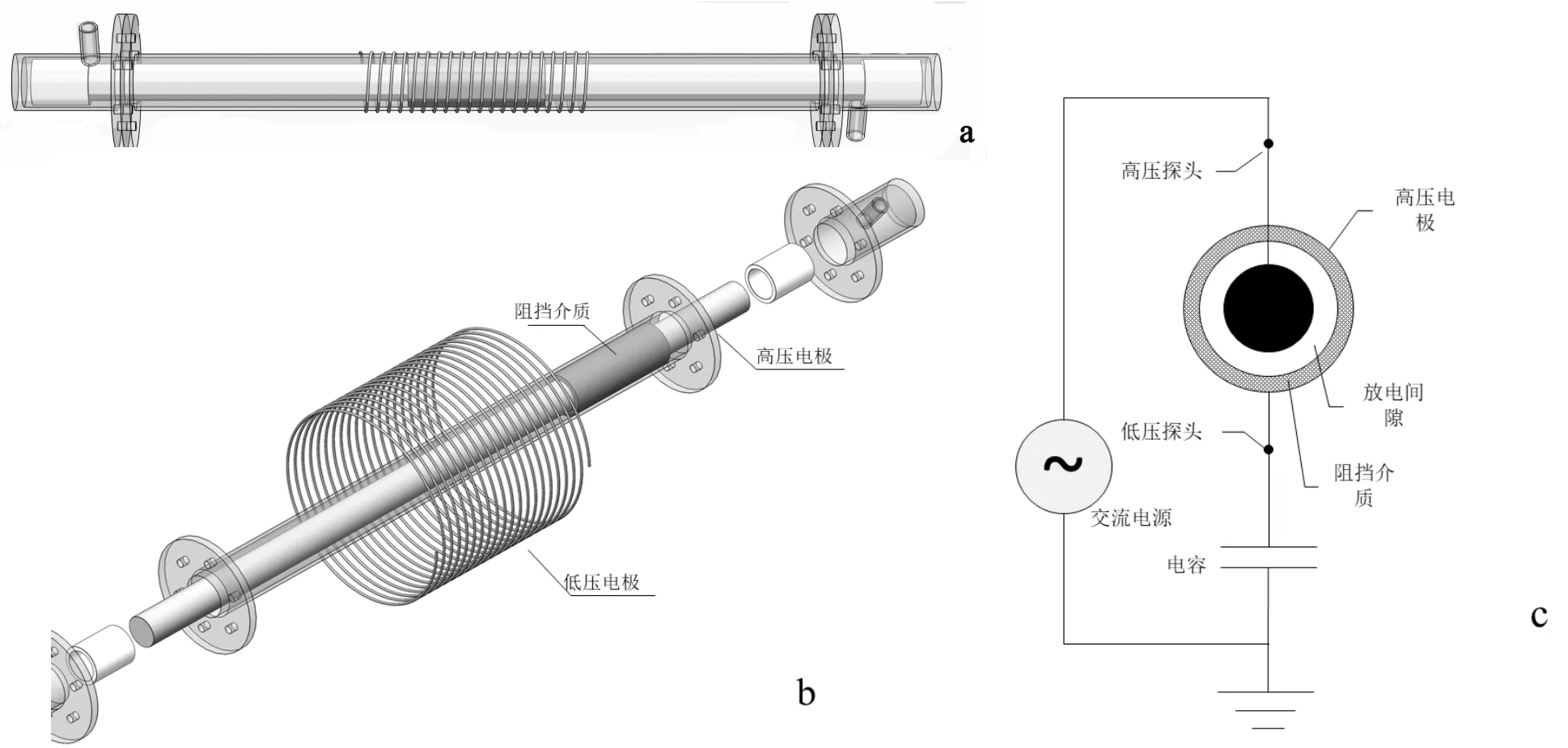
图2 DBD反应器结构示意
a: 整体外观;b:每个组件的分解图;c:测量位置
废气检测与后处理系统:使用烟气分析仪(带有CO,SO2,O2和N2传感器的ECOM-J2KN)检测废气中的SO2浓度.使用臭氧监测仪(2B-205)分析O3的浓度.首先将3个处理过的废气样品(未处理对照组、仅用催化剂处理组、串联DBD反应器增强的催化剂协同处理组)收集到10L气囊中,然后泵入烟气分析仪中.在确定每个废气样品中的SO2浓度之后,使用公式(1)计算脱硫效率:
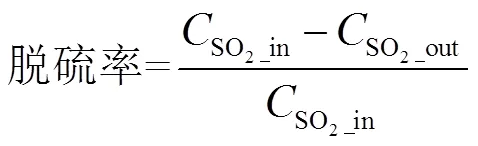
1.3 实验流程
预硫化:配制N2、CO和SO2体积比为137: 10.5:17.5(模拟电解铝烟气配比)的烟气,在催化剂活性评价系统中于600℃下对制备好的催化剂进行6h的预硫化.所有催化剂在使用前均经过预硫化操作.
通入典型电解铝烟气:预硫化结束后,利用质量流量计控制不同钢瓶气的流速,以使得混合气体组成与典型电解铝烟气组成基本相符,以1L/min的流量通入后续的各系统中.
进行催化剂活性评价:将前述制备的各组催化剂填装入活性评价系统中,在不同的催化反应温度下仅使用催化剂进行催化脱硫.
进行强化脱硫:控制DBD反应器的放电功率,固定注入能量密度.使烟气获得充分激发,使激发后的烟气在不同的催化反应温度下通过催化剂评价系统进行脱硫.
测定降解效率:通过烟气分析仪对处理后的尾气进行分析以获得SO2的降解效率.
1.4 催化剂表征
1.4.1 X射线衍射(XRD) 采用Bruker D8ADVANCE A25X对催化剂进行X射线衍射实验,在束电压为40kV的Cu辐射下,范围为10°~90°,速度为6°/min.
1.4.2 扫描电子显微镜(SEM) 采用NOVANANOSEM-450扫描电子显微镜(SEM),通过能量色散X射线光谱法(EDS)来强化对催化剂表面结构的观察和分析,加速电压为30kV.
1.4.3 氮的吸附和解吸 采用表面积孔径分析仪(3H-2000PM2,贝塞斯达仪器技术(北京)有限公司)通过氮气吸附和解吸实验以及BET法评估了催化剂的比表面积和孔体积.样品在200℃下脱气120min,氮气为吸附物.氮气工作温度为77.3K,饱和蒸气压为0.099MPa.使用Langmuir方法确定比表面积,并通过Barrett-Joyner-Halenda(BJH)方法(圆柱孔模型,1.9~42.1nm)确定孔体积和直径.
1.4.4 X射线光电子能谱(XPS) 采用Thermo Fisher Esca Lab 250Xi仪器上使用单色Al K源(光子=1486.6eV)在10mA灯丝电流和14.7keV灯丝电压下进行的,实验在0.5mm的场中通过能量为30eV.电荷中和剂用于补偿样品的任何电荷,使用284.8eV的脂肪族C1s峰的结合能对光谱进行了校准.
2 结果与讨论
2.1 DBD反应器表征
为了避免能量浪费以及副产物的产生,将输入能量控制在刚刚产生细丝放电的最低限制,放电频率固定在9.4kHz即每个放电周期内可以产生6~7次细丝放电,可以认为整个电流通道充满了均匀放电的细丝,可认为将1L/min的模拟烟气通入放电装置时每个气体分子受到激发的概率相等.此时产生的-图和李萨如图及放电照片如图3所示.
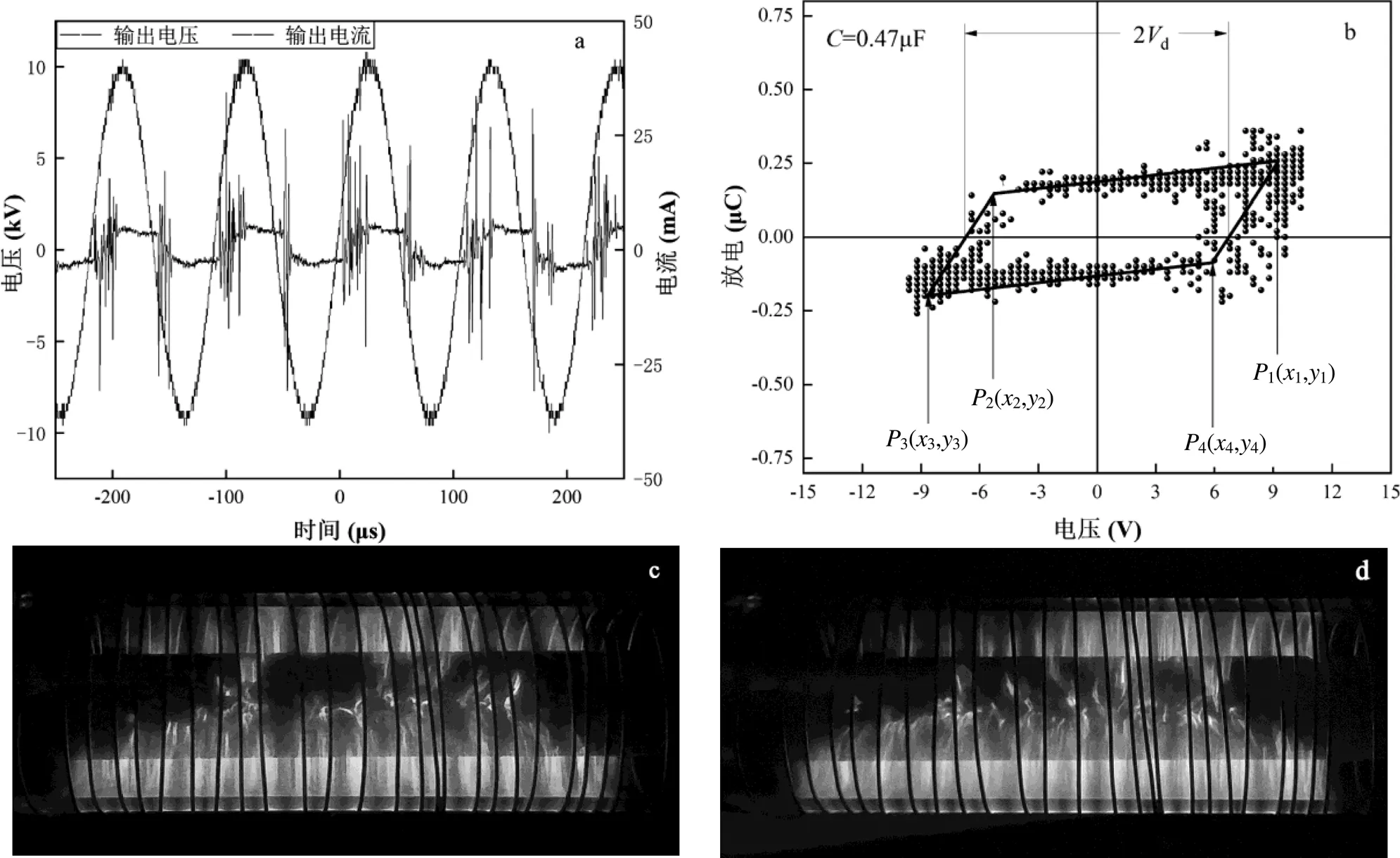
图3 DBD放电特征
a 电流-电压-时间曲线;b李萨茹图形;c等离子体低输出模式摄影图像;d等离子体高输出模式摄影图像
首先使用李萨如图面积积分法计算DBD反应器的能量密度,DBD反应器参数在有限的输出模式是:总负载电容=6.01pF、等效介质电容=49.8pF、等效电容的放电间隙=6.834pF、气隙电压= 6710V、放电功率=19.989W.流量()固定在1L/min.由于各组实验的脱硫效率均于气流量、放电条件一致的情况下测得,因此对于所有实验组,放电条件没有发生改变.则各实验组的指定输入能量均可用公式(2)进行计算:
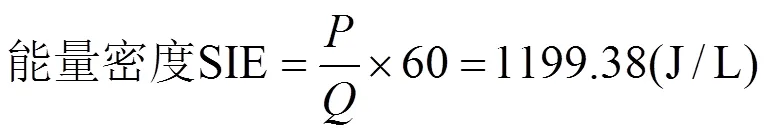
从计算结果可以看出,DBD反应器是在低功率状态下运行,因此反应器整体的升温现象不明显,可以长时间稳定运行.由于反应器的气隙电容偏小,使得反应器输出功率受限.若需进一步提升等离子体反应器功率,增加脱硫效率,除了增加放电电压外,也可以采用减小气隙的方式.该方法可有效提高同电压下等离子体反应器的能量密度,更易产生大量的高能粒子.但是盲目增加反应器的能量密度会造成能量的浪费.
用积分法计算李萨如图形面积:
图3b中P3P4线段的斜率给出了电源的总负载电容t:
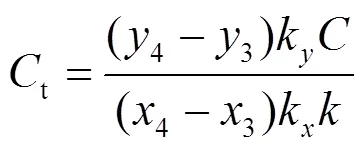
由P1P4线的斜率可得绝缘介质放电时的等效介电电容d:
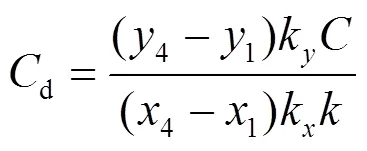
放电间隙的等效电容为:
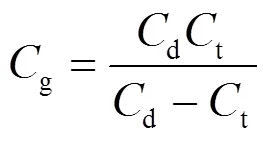
气隙放电时的电压d由P2P3线与P4P1线在轴上的截距可得:
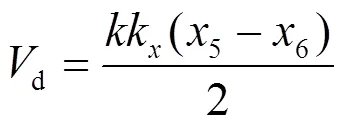
李萨如模式(平行四边形)中的面积为放电功率:
=´´´k´k´(7)
式中:是内部采样电容的电源,是0.47mF.电压采样比为1000:1(电压的实际坐标单位为kV).CH1和CH2的信号采集衰减倍数分别为k和k=1(即本实验中没有衰减).
从图3c~d的放电图像可以看出,随着能量密度的增加,放电的颜色愈加明亮.这表明单根细丝所携带的能量急剧增大,使等离子体中的电子携带过量的能量,当电子与气体分子间进行能量传递之后,携带过量能量的电子为释放能量,就会与反应器的器壁进行激烈的碰撞,造成反应器升温,运行寿命大大降低.除此之外,被过度激发至高能态的亚稳态粒子在离开放电区域之后为释放能量会进行副反应,产生大量副产物,使烟气脱硫困难.
2.2 脱硫率
2.2.1 单催化剂脱硫率 如图4所示,Cu负载量5%的催化剂脱硫率最差,直到500℃附近才表现出部分催化活性.其次是Cu负载量为10%的催化剂,其脱硫率随着温度的上升而上升,效率最高达到55%.效果最好的一组为Cu负载量为20%的催化剂.其脱硫率在各个温度段都要超过5%及10%负载量的催化剂,效率最高达到82.3%.此外,由图中还可看出,催化剂制备时的老化温度也会对脱硫率造成较大影响,40℃的效果最佳.因此往后实验采用含铜量为20%,老化温度为40℃的催化剂.
2.2.2 等离子体诱导脱硫率提高 由图5可知,等离子体强化后各催化剂组的脱硫率均比单独催化剂组的脱硫率有不同程度的提高.在催化剂中,掺杂20%铜的3组催化剂效率最高,Cu负载量为20%,老化温度为40℃,反应温度为450℃下反应的催化剂的脱硫率为81.8%,而在没有等离子体的情况下为51.2%.其余各组都有不同幅度的提升,其中,Cu负载量为20%,反应温度为500℃,老化温度为40℃的催化剂在强化后脱硫效率已达到100%(未强化前82%).由此可见,DBD放电对20%铜掺杂催化剂的影响最为显著,这是因为负载20%Cu的催化剂晶胞整体禁带宽度最低,因此最容易被激发.使得在有额外能量供给的条件下,各种能级的能量都能充分被催化剂吸收利用,使得催化剂上的电子空穴数量远超另外两种掺杂比例的催化剂.因此宏观上来看DBD对该组催化剂的强化效果最好.因此利用DBD反应器进行强化催化可在催化效果不变的前提下使催化温度得到大幅降低.
图6a可以看出,在DBD反应器刚刚产生细丝放电时的能量密度为1199.38J/L,此时3组催化剂降解效率均未超过50%,随着反应器功率的上升,脱硫率急剧上升.当反应器功率为原来的1.5倍时,含铜20%的催化剂已经接近100%,随着功率的进一步上升,当能量密度达到2000J/L时3组催化剂均达到100%的降解率.
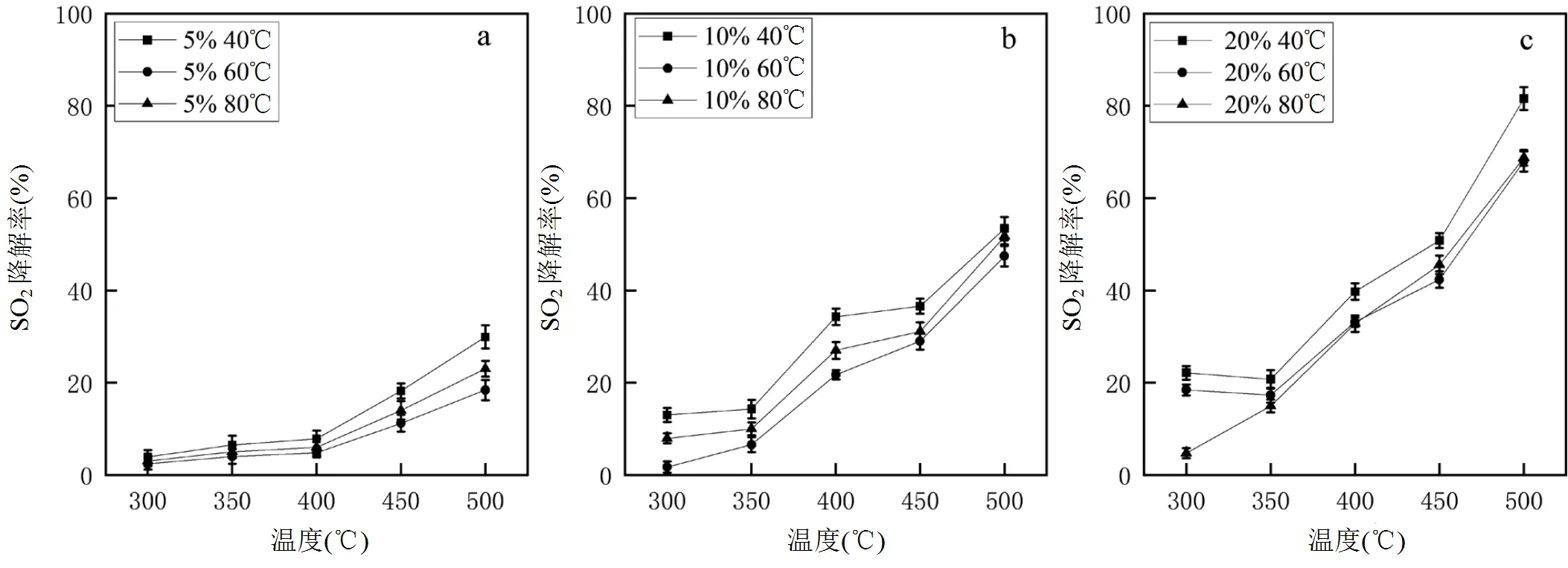
图4 各种温度下老化的Cu-Sn-Zr催化剂的SO2去除效率

图5 不同催化剂在DBD增强前后脱硫率对比
a.单独催化剂;b.催化剂+等离子体
CO和O3都是该反应的副产物,以O3为例分析催化剂对副产物的抑制作用.从图6b可以看出,随着能量密度的上升,O3的生成量也在逐渐上升.掺杂20%Cu的催化剂的O3生成速率相当低,与单独等离子体相比抑制了95%O3的生成.而掺杂5%Cu的催化剂在能量密度约为800J / L时,O3的生成速率急剧增加,这意味着DBD提供的能量可以被20%Cu掺杂的催化剂利用,而Cu负载较低时能量利用率低;当提供过量能量时,它主要用于促进催化剂能带之间的电子跃迁,从而避免了能量浪费.但是对于5%Cu掺杂的催化剂,禁带较宽会阻碍一定水平能量向分子气体的有效转移.而达到一定量的电子浓度后过量的能量对反应正向进行不再有积极的促进作用,反而会使其沿着放电通道与反应器壁激烈碰撞,使电子能量转化为热能,造成反应器的急剧升温,同时造成严重的能量浪费.同时也会产生大量的副产物,给后续处理带来困难.
2.2.3 脱硫机理 传统SO2的还原过程涉及双重催化反应机理.即氧化还原反应和COS中间机理.具体的反应路径偏好取决于催化剂的活性种类,两个反应可以同时发生或分别发生.因此收集20%Cu负载量的催化剂COS选择性数据,根据式(8)计算COS的选择性,并绘制图7.
由于反应生成的S数量少,并且冷凝在DBD反应器后段的内表面,难以收集分析.因此检测COS来分析脱硫反应的选择性.

从图7中可以看出,随着温度的升高,对COS的选择性逐渐降低.表明更多的COS参与了克劳斯反应,导致减少了更多的SO2.放电后,对COS的选择性进一步降低.克劳斯反应的一部分活化能可以由等离子体提供,使得该反应可以在较低温度下发生.
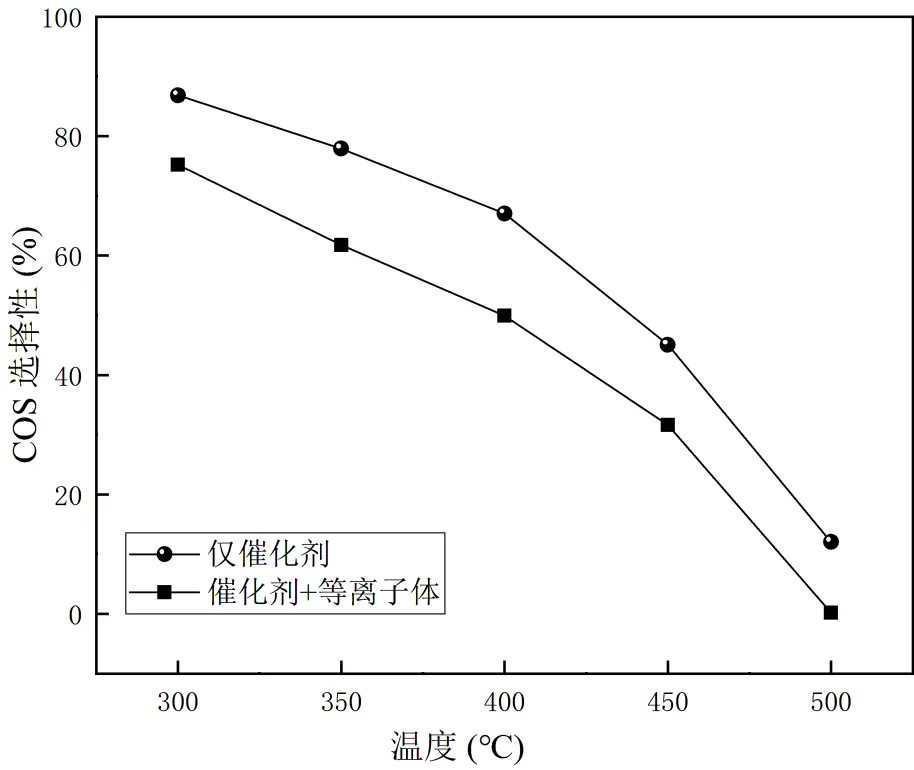
图7 20%Cu负载Sn-Zr催化剂在等离子体辅助下COS选择性与温度的关系
2.3 催化剂表征
2.3.1 XRD表征 对Cu负载为20%,老化温度为40℃的催化剂进行了XRD表征,考察其在刚制备完成时,预硫化完成后以及在等离子体强化条件下使用1h后的晶型,如图8所示.
新鲜的催化剂在2=26.4°、33.8°、51.7°等位置出现了明显的SnO2的特征峰,在35.5°、38.6°、67.9°等位置出现了部分CuO的特征峰.但是与ZrO2的主要特征峰没有明显的对应.可以认为Zr元素在催化剂中是以表面分散相的形式存在,并未参与结晶.说明在催化剂中构成主要晶相的为SnO2,Cu元素参与成晶,但是不构成主晶相. Cu以杂原子掺杂的形式替代SnO2晶胞结构中的Sn元素.因此证明后文中数值分析中晶胞模型构建时忽略Zr成晶的正确性.另外,由于三者之间的晶型没有明显差异,放电并没有改变催化剂的晶体结构,产生催化活性的主要晶相也没有改变.
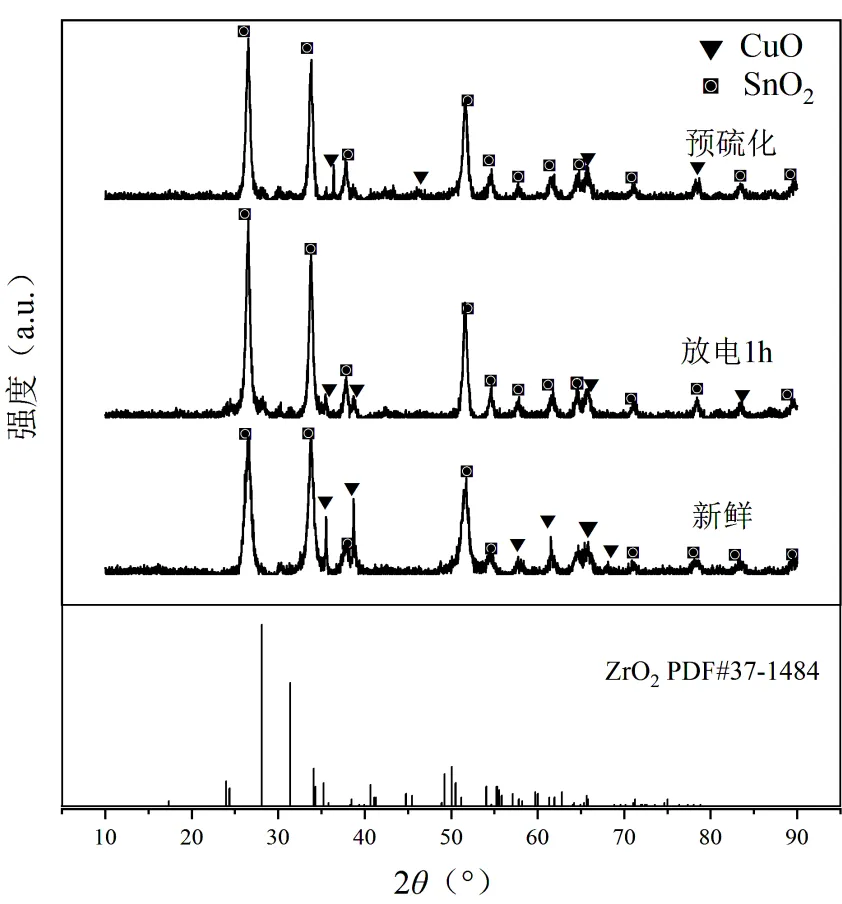
图8 20%-Cu掺杂(40℃老化)催化剂在各种条件下的XRD
2.3.2 SEM和EDS表征 对同一组催化剂(Cu负载量为20%,老化温度为40℃)进行了SEM表征,并对其表面进行了EDS线扫(图10).
由图9a、b中可看出,新鲜催化剂中形成了较为明显的颗粒状结构.在晶粒的每个表面上又会附着一部分未完全成晶的催化剂颗粒.
由图9c、d可看出经过预硫化后的催化剂颗粒表面出现明显的表面熔融烧结,颗粒分散情况下降,烧结可能使得催化剂内部孔道被堵塞.虽然预硫化所产生金属硫化物能够显著提升COS中间体反应,但是由于催化剂的表面孔道被堵塞,使得活性位点难以接触反应物,脱硫性能并没有明显提高.而且随着催化剂使用次数的增加,反应所产生的S元素堵塞催化剂表面和孔道,使得孔容下降,使脱硫效率上升较少.

图9 负载20%铜的催化剂在不同条件下的SEM图像
a、b新鲜;c、d预硫化;e、f放电1h
由图9e、f可以看出,放电后,催化剂在等离子体所产生的高能粒子持续照射下产生了类似刻蚀的效果,表面的沉积物得到有效的清除,烧结情况也得到明显改善,随着表面熔融层的去除,催化剂的孔道结构得以暴露出来,使得催化剂的比表面积和孔容都得到了提升(与BET表征的数据相吻合).从而让催化剂内表面获得了高效的利用,使得催化效率得到极大的提升.另外,长时间高能粒子的撞击将之前较规则的大颗粒打碎,使得催化剂颗粒尺寸更加均匀,降低了内扩散的影响.因此可以使催化剂在使用过程中得到同步再生,使催化剂的使用寿命得到明显增加.
如图10a所示,新鲜催化剂中仅检出Cu、Zr、Sn、O 4种元素的峰,而预硫化过后出现了明显的S的峰.由脱硫反应路径可知,预硫化的主要目的是生成金属硫化物,从而提高COS中间体反应在催化反应中的占比.但是预硫化金属氧化物催化剂在反复使用后容易出现金属硫化物重新变为金属氧化物,反应路径又重新偏向于Redox反应的情况,这使得脱硫效率随着催化剂的使用次数增加而降低.而在本实验中,BDB处理过后的催化剂中的金属硫化物在多次使用后依然稳定存在,对维持COS中间体反应起到积极作用.
由图10b可知,新鲜催化剂中没有S元素,但是Sn元素占比较高,可以认为表面主要形成了Sn的结晶且大量Sn处于催化剂表面,经过预硫化后, 开始出现S元素,O和Sn的含量均稍有下降,可以认为催化剂表面由于烧结使得晶粒中的Sn被熔融物所包裹.而在经过长时间高能粒子冲击过后,可以看到S的含量不降反升,一方面可以认为是还原过后的S单质在催化剂表面的沉积,另一方面也可认为是活性粒子长时间撞击催化剂表面,使得S以某种循环的方式维持住金属硫化物的特性,从而延长了催化剂寿命.同时O与Cu的含量也有提升.可以认为原本的催化剂大颗粒结构在放电后被打碎,使得原本包覆在颗粒内部的Cu得以暴露出来,从而让3种有效成分得以充分参与反应,最终使催化反应效率上升.
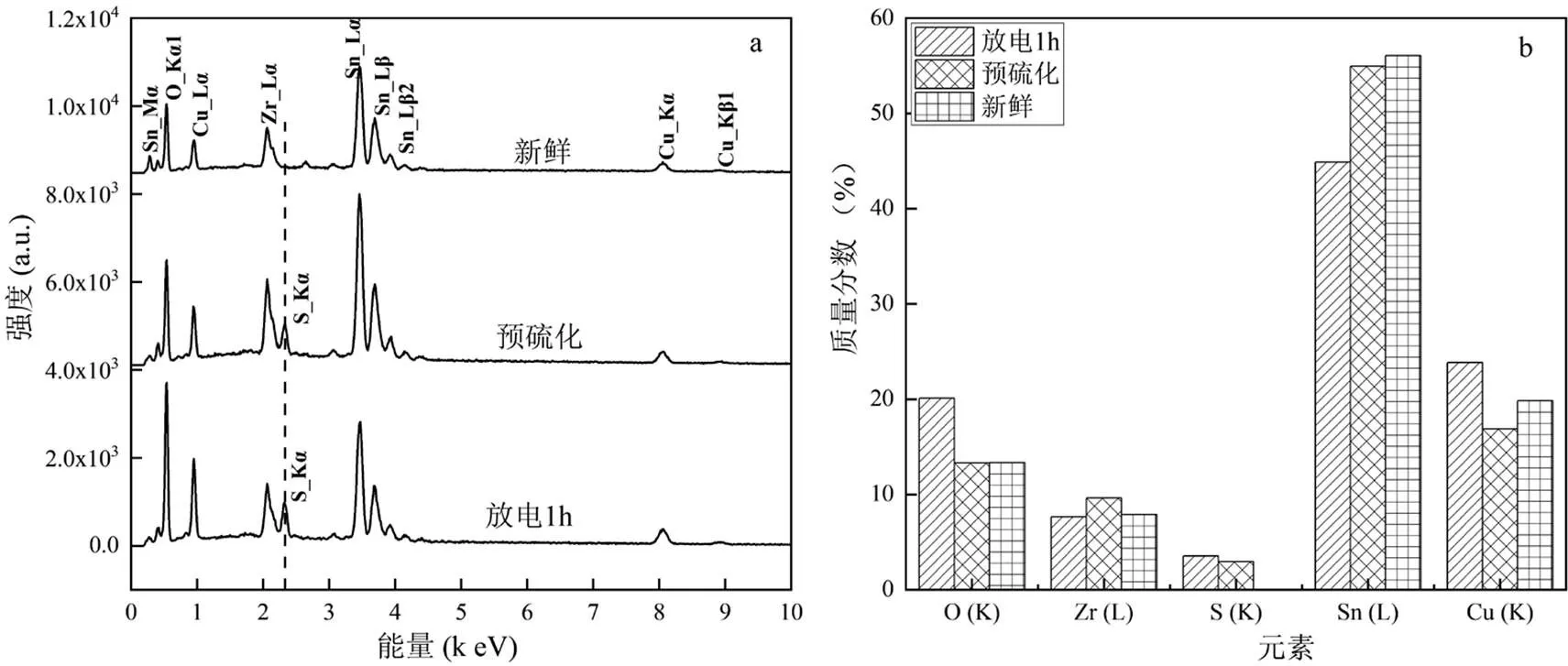
图10 三种催化剂(新鲜,预硫化,放电1h)的能谱和元素重量比
K,L是元素的特征X射线的线系
2.3.3 N2-物理吸附 对Cu负载量为20%,老化温度为40℃的催化剂进行N2-物理吸脱附测试,如图11所示.吸附为型等温线,证明吸附质分子间的相互作用比吸附质与吸附剂之间的强,第一层的吸附热比吸附质的液化热小.经过放电后的催化剂吸脱附等温线中的回滞环明显增大,且吸附容量上升.另外,采用Langmuir法测试结果比表面积为29.07m2/g,而预硫化后放电40min催化剂比表面积增加至48.71m2/g,扩大了19.64m2/g,增长率达67.56%.使用BJH脱附法所测的孔体积由处理前的0.1137mL/g增加至0.1488mL/g,扩大了0.0351mL/g,增长率为30.87%.同样使用BJH吸附法所测量得到的最可几孔直径由处理前的38.29nm减小为2.67nm,减少93.03%.但结合SEM照片的结论可推测,该催化剂形成的主要为晶粒而非晶胞.
结论也与降解效率曲线相一致,改性过后的催化剂由于等离子体产生的高能粒子持续撞击,使得催化剂表面产生类似刻蚀的效果,比表面积扩大,一方面使得催化剂孔道中的活性位点更易暴露,另一方面孔容上升,同时期吸附的反应物分子上升,使得催化效率上升.
由图11也可看出,新鲜催化剂的d/d较大的位置集中在20nm上下,而其他粒径段的孔分布较为均匀,整体孔容也较低.而放电40min后的催化剂整体的d/d都有所上升,其中20nm及3nm左右的d/d上升明显.证明改性方案对催化剂孔道结构有明显的调控作用,尤其对3nm~20nm孔径段的孔有着明显的促进作用.
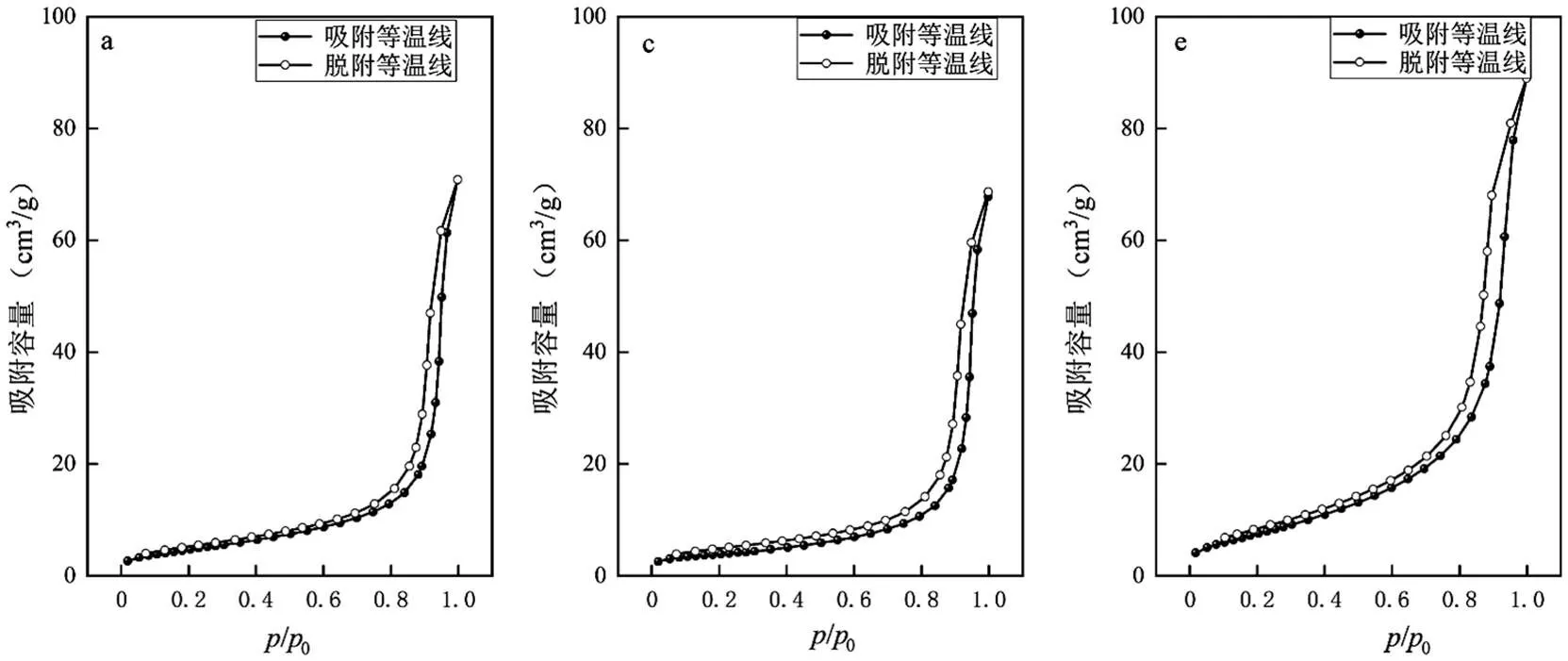
a、b新鲜催化剂;c、d预硫化催化剂;e、f放电 1h后催化剂
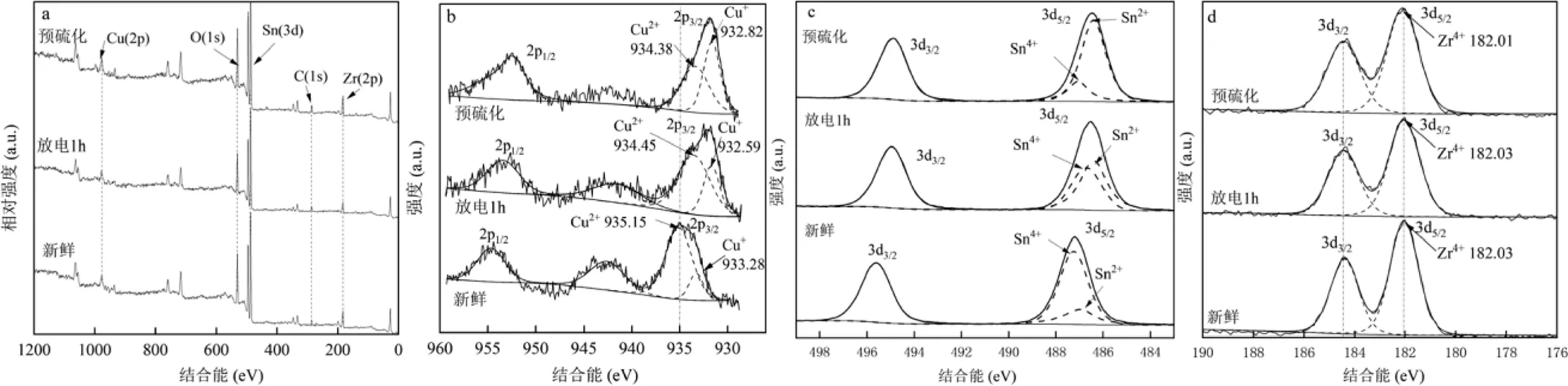
图12 不同处理催化剂的XPS谱图
a: 全谱;b: Cu 2p;c: Sn 3d ;d: Zr 3d
2.3.4 XPS表征 对Cu负载量为20%,老化温度为40℃的催化剂进行XPS表征,结果如图12 所示.经过预硫化后的Cu(2p)轨道的峰出现了明显的向高场偏移,部分Cu1+转变为Cu2+,说明Cu失去电子,也就是说形成催化剂后,Cu上的电子会部分转移给原有催化剂,从而使原有的金属氧化物形成更多的金属硫化物.另外可以注意到,在持续放电1h后该偏移依然存在.对于Sn元素来说,新鲜催化剂表面主要以SnO2(+4)的结晶态存在,预硫化过后部分被还原成低价态氧化物或者硫化物参与COS中间体反应和生成多的晶格氧空位.在放电1h过后,金属硫化物催化剂被CO消耗生成COS,参与到COS反应机制中去,导致+2价比例下降.同时,等离子体反应器中存在着O2和O3,+2价的部分Sn被氧化成高价,晶格氧空位消失.
2.4 数值分析
放电会使催化剂的物化特性发生变化,进而影响其选择性.就放电而言,若在放电空间内填入催化剂,则放电将由单纯的细丝放电转变为催化剂颗粒间/颗粒内部的表面爬电和微孔放电(催化剂颗粒接触点上形成的高电场),从而使平均电子能量增加,进而加强了离子化过程.而催化剂的能带结构能直接反映其离子化过程与NTP强化催化,因此对催化剂性能进行理论计算,解析其能带结构.利用Materials Studio软件中的CASTEP计算包对催化剂的态密度及能带结构进行了计算.
计算过程中,催化剂单元统一采用2´2´2的超晶胞模型进行构建,基础晶格单元为SnO2.由XRD结果可以判断,Zr原子没有特征峰,因此可以认为Zr元素在催化剂中呈分散相,没有参与成晶.因此在催化剂晶胞构建过程中没有添加Zr元素.构建结束后超晶胞中共含有48个原子.随后使用Matlab中modAtom.p程序对Materials Studio生成的模型进行更改以完成晶胞中Cu元素的替换.替换的原子个数不同,对应的掺杂浓度为也就不同.理论模拟计算中,分别使用替换1, 3, 5个元素的方式来模拟实际催化剂制备时5%,10%,20%三个掺杂浓度.
SnO2的超晶胞经过优化以后,计算之后得到其晶格常数和晶胞体积分别为=4.695Å,=3.162Å和=69.73Å,Pianaro等[25]研究组实验中所测得的晶格常数为=4.737Å,=3.185Å和=71.49Å,所以理论计算和实验测试所得到的数据基本保持一致.Cu掺杂SnO2的超晶胞结构经过优化之后,晶格常数和晶胞体积分别为=4.701Å,=3.17Å和=70.05Å3. Mäki- Jaskari[26]研究组利用超软赝势法对直接化学配比的SnO2进行了计算,其结果表明,纯SnO2是一个直接带隙半导体,导带的最低点与价带的最高点均位于布里渊区点的Г点.计算获得的带隙宽度为1.2eV,能带结构结果表明SnO2具有半导体的性质.在此基础上,3种不同Cu含量掺杂后的态密度图如图13所示.
由图13中可以看出,随着Cu掺杂数量上升,催化体系的禁带宽度逐渐降低.Cu的负载量为5%,10%和20%的催化剂其催化体系的禁带宽度分别为5.99 ,5.73和5.26eV.当Cu掺杂量为5%时电子在各个能级上出现的概率较为集中,各电子轨道上的概率密度峰较高.这意味着处于基态的电子需要较高的能量才能获得激发,该种催化剂对低级能量的利用率最差.当Cu掺杂量上升至10%时,电子概率密度有向各个能级平均分配的趋势,意味着电子在能级间跃迁能力有所上升.但是由于0eV处概率密度再次偏向负电位区域,导致禁带宽度没有大幅下降,整体来说该种催化剂对能量的利用效率有所上升,但提升有限.而当Cu掺杂达20%时,可以看到在0eV处出现了一个较为明显的肩峰,4eV处的峰也向下移,使得整体的禁带宽度只有2eV,同时高能级态下各电子轨道上的概率密度分布更加均匀,意味着电子在各级轨道中迁移要更加容易,因此可以认为该材料在不同能量的激发下都能产生相应的电子激发,从而更加容易产生电子空穴,使催化剂对低级能量的利用效率上升.
因此,可以认为,随着Cu的掺杂量的增加,整个复合催化剂的能带结构在发生变化,主要表现在价带的宽度增加,导带下移,最终导致了整个禁带宽度的减小.当Cu的掺杂量到20%附近时,整个复合催化物的禁带宽度已经接近0eV.再继续增加Cu的掺杂量,一方面可能破坏SnO2体系半导体的性质,另一方面会使得体系中Sn含量下降,破坏原有的活性位点.所以单纯的通过能带理论的计算,可以认为当掺杂量为20%时,整个催化体系的催化效果会达到最好.
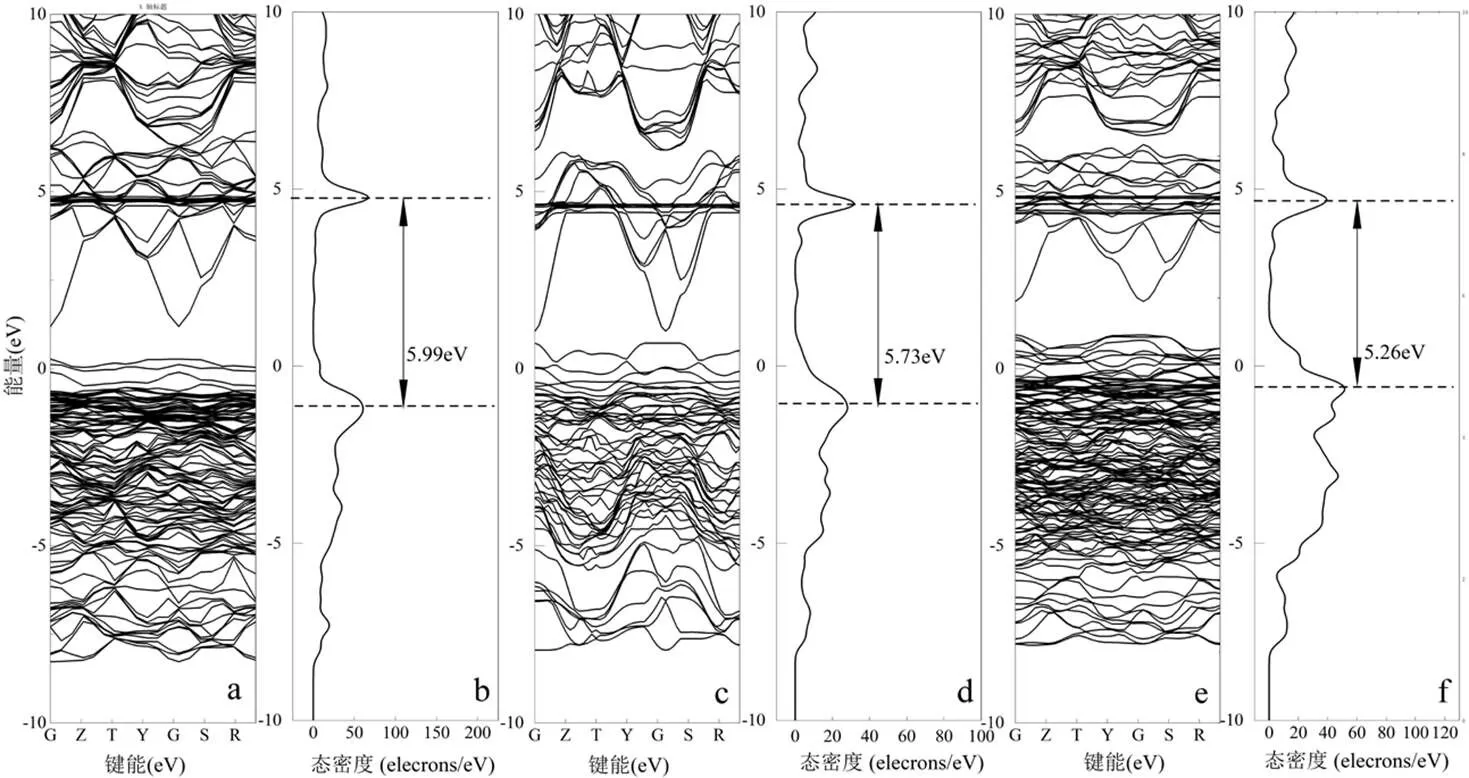
图13 不同Cu掺杂比的Cu-Sn-Zr催化剂的能带结构和态密度
(a, b) 5%Cu掺杂; (c, d) 10%Cu掺杂; (e, f) 20%Cu掺杂
GZTYGSR为布里渊区高对称点
3 结论
3.1 掺杂20%铜的3组催化剂效率最高,其中,Cu负载量为20%,反应温度为500℃,老化温度为40℃的催化剂在强化后脱硫效率已达到100%(未强化前82%). DBD放电对20%铜掺杂催化剂的影响最为显著.
3.2 掺杂20%Cu的催化剂的O3生成速率与单独等离子体相比抑制了95% O3的生成.而掺杂5%Cu的催化剂能量密度约为800J/L时,O3的生成速率急剧增加,这意味着DBD提供的能量可以被20%Cu掺杂的催化剂高效利用,而Cu负载较低时能量利用率低;当提供过量能量时,它主要用于促进催化剂能带之间的电子跃迁,避免了能量浪费.但是对于5%Cu掺杂的催化剂,禁带较宽会阻碍一定水平能量向分子气体的有效转移.
3.3 三元催化剂中的主要晶体结构为SnO2,CuO部分结晶.随着Cu掺杂数量上升,催化体系的禁带宽度逐渐降低.Cu负载量为5%,10%和20%的催化剂体系的禁带宽度分别为5.99,5.73和5.26eV.
[1] Hanif M A, Ibrahim N, Jalil A A. Sulfur dioxide removal: An overview of regenerative flue gas desulfurization and factors affecting desulfurization capacity and sorbent regeneration [J]. Environmental Science and Pollution Research, 2020,22(27):27515–40.
[2] 余 创,彭学斌,田 林,等.电解铝烟气脱硫技术研究进展 [J]. 云南冶金, 2019,4(48):40-43.
Yu C, Peng X B, Tian L, et al. Research progress of electrolytic aluminum flue gas desulfurization technology [J]. Yunnan Metallurgy, 2019,4(48):40-43.
[3] Wang X H, Wang A Q, Li N, et al. Catalytic reduction of SO2with CO over supported iron catalysts [J]. Industrial & Engineering Chemistry Research, 2006,45(13):4582-4588.
[4] Feng T, Zhao X, Wang T, et al. Reduction of SO2with CO to elemental sulfur in activated carbon bed [J]. Energy & Fuels, 2016,30(8): 6578-6584.
[5] Han G B, Park N K, Yoon S H, et al. Synergistic catalysis effect in SO2reduction by CO over Sn–Zr-based catalysts [J]. Applied Catalysis A General, 2008,337(1):29-38.
[6] Han G B, Park N K, Lee T J. Effect of O2on SO2reduction with CO or H2over SnO2-ZrO2Catalyst [J]. Industrial & Engineering Chemistry Research, 2009,48(23):10307-10313.
[7] Park N K, Park J Y, Lee T J, et al. Catalytic reduction of SO2over Sn-Zr based catalysts for DSRP under high pressure [J]. Catalysis Today, 2011,174(1):46-53.
[8] Park N K, Lee T H, Lee T J, et al. Catalytic reduction of SO2under the regeneration of off-gas containing oxygen over Cu-Sn-Zr-based oxides for the hot coal gas desulfurization process [J]. Catalysis Today, 2016,265:131-137.
[9] Happel M, Lykhach Y, Tsud N, et al. SO2decomposition on Pt/CeO2(111) model catalysts: On the reaction mechanism and the influence of H2and CO [J]. The Journal of Physical Chemistry C, 2012,116(20):10959-1096.
[10] Zhao H, Luo X, He J, et al. Recovery of elemental sulphur via selective catalytic reduction of SO2over sulphided CoMo/γ-Al2O3catalysts [J]. Fuel, 2015,147(may 1):67-75.
[11] Gao G P, Wei S H, Guan X M, et al. Influence of charge state on catalytic properties of PtAu(CO)n in reduction of SO2by CO [J]. Chemical Physics Letters, 2015,625:128-131.
[12] Zhang L, Qin Y H, Chen B Z, et al. Catalytic reduction of SO2by CO over CeO2–TiO2mixed oxides [J]. Transactions of Nonferrous Metals Society of China, 2016,26(11):2960-2965.
[13] Ban, Z, Zhang, J, Wang, S, et al. Direct reduction of SO2to elemental sulfur by the coupling of cold plasma and catalyst (I) [J]. Industrial & Engineering Chemistry Research, 2004,17(43):5000-5005.
[14] Mizuno A, Clements J S, Davis R H. A method for the removal of sulfur dioxide from exhaust gas utilizing pulsed streamer corona for electron energization [J]. IEEE Transactions on Industry Applications, 2008,22(3):516-522.
[15] Onda K, Kasuga Y, Kato K. Electric discharge removal of SO2and NOfrom combustion flue gas by pulsed corona discharge [J]. Energy Conversion & Management, 1997,38(10):1377-1387.
[16] Feng F, Ye L, Liu J, et al. Non-thermal plasma generation by using back corona discharge on catalyst [J]. Journal of Electrostatics, 2013, 71(3):179-184.
[17] Feng F, Zheng Y, Shen X, et al. Characteristics of back corona discharge in a honeycomb catalyst and its application for treatment of volatile organic compounds [J]. Environmental ence & Technology, 2015,49(11):6831-6837.
[18] Moon J D, Jung J S, Effective corona discharge and ozone generation from a wire-plate discharge system with a slit dielectric barrier [J]. Journal of Electrostatics, 2007,65(10/11):660-666.
[19] Mista W, Kacprzyk R. Decomposition of toluene using non-thermal plasma reactor at room temperature [J]. Catalysis Today, 2008, 137(2-4):345-349.
[20] Moscosa-Santillan M, Vincent A, Santirso E, et al. Design of a DBD wire-cylinder reactor for NOemission control: experimental and modelling approach [J]. Journal of Cleaner Production, 2008,16(2): 198-207.
[21] Schiorlin M, Marotta E, Rea M, et al. Comparison of toluene removal in air at atmospheric conditions by different corona discharges [J]. Environmental science & Technology, 2009,43(24):9386-9392.
[22] Ban L, Liu P, Ma C, et al. Deep desulfurization of diesel fuels with plasma/air as oxidizing medium,diperiodatocuprate (III) as catalyzer and ionic liquid as extraction solvent [J]. Plasma science and Technology, 2013,15(12):1226-1231.
[23] Ban L L, Liu P, Ma C H, et al. Deep oxidative/adsorptive desulfurization of model diesel oil by DBD/FeCl3–SiO2[J]. Catalysis Today, 2013,211:78-83.
[24] Zhang H, Wang W, Li X, et al. Plasma activation of methane for hydrogen production in a N2rotating gliding arc warm plasma: A chemical kinetics study [J]. Chemical Engineering Journal, 2018,345: 67–78.
[25] Pianaro S A, Bueno P R, Longo E, et al. Microstructure and electric properties of a SnO2based varistor [J]. Ceramics International, 1999,25(1):1-6.
[26] Maki-Jaskari M A, Rantala T T. Density functional study of Pd adsorbates at SnO2(110) surfaces [J]. Surface Science, 2003,537(1-3): 168-178.
致谢:感谢云南大学国家实验化学与化学工程教育示范中心提供的技术帮助.感谢云南大学化工学院郝天辉、王则月、朱忍和胡然同学在实验中的帮助,感谢各位老师在实验中的规划和指导.
The characteristics of CuSnZr catalyst applied to NTP enhanced catalytic desulfurization process.
ZHOU Zheng-hua1, JIANG Lian-shuang2, ZHANG Zhen-yu2, HUANG Rui2, NING Jing-yuan3, NING Zhi-yuan2*
(1.School of Materials and Energy, Yunnan University, Kunming 650000, China;2.School of Chemical Science and Technology, Yunnan University, Kunming 650000, China;3.Kunming Zhongkeyun Environmental Protection Co. LTD, Kunming 650000, China)., 2022,42(1):20~31
In order to efficiently process a large amount of low-concentration electrolytic aluminium flue gas at low temperature, the Sn Zr type metal oxide was prepared by the impregnation method with Cu added as a catalyst, and this desulfurization effect of CuSnZr catalyst was the first time to be tested in low temperature plasma (NTP) technology. The best desulfurization performance results when the catalyst was loaded with 20wt% Cu and when the aging temperature was 40℃. Compared with the fresh catalyst, the X-ray diffraction analysis (XRD) results showed that the discharge basically did not affect the crystal form of the catalyst; the scanning electron microscopy (SEM), nitrogen adsorption and desorption (BET) indicated that the discharge will greatly improve the adsorption capacity, the desorption capacity and the pore structure of the catalyst; X-ray photoelectron spectroscopy (XPS) also showed that the discharge will change the valence state of the elements on the catalyst surface, thereby changing its redox performance and deviating the reaction path. For the catalyst performance analysis, theoretical calculations indicate that the increase in copper content will cause changes in the energy band structure of the catalyst, which is better used for exciting gas.
catalyst;plasma-enhanced catalytic;aluminium flue gas desulfurization;DBD discharge;preparation;numerical analysis
X511
A
1000-6923(2022)01-0020-12
周正华(1996-),男,云南大理人,云南大学硕士研究生,主要从事脱硫催化剂材料研究.
2021-05-10
云南省科学技术厅科学技术计划(2018FD013)
* 责任作者, 讲师, zhiyuan_ning@outlook.co
猜你喜欢
弹性体(2022年3期)2022-11-15
轮胎工业(2022年2期)2022-07-19
石油化工高等学校学报(2022年1期)2022-04-15
烟台果树(2021年3期)2021-07-21
轮胎工业(2021年1期)2021-07-19
空间科学学报(2021年6期)2021-03-09
中外葡萄与葡萄酒(2019年2期)2019-04-12
上海农业科技(2019年1期)2019-02-22
北京航空航天大学学报(2017年7期)2017-11-24
北京航空航天大学学报(2017年7期)2017-11-24
