
特殊的慢性粒细胞白血病一例
2022-01-12 13:13张绪湃陈柯材
上海医学 2021年12期
张绪湃 曾 洁 徐 娅 程 红 陈柯材 张 婷
1 临床资料患者男,53岁,因“体检发现白细胞及血小板计数升高”于2020年12月14日入住内江市第一人民医院血液科。患者入院前半天于当地医院体检,血常规示白细胞计数113.98×109/L、血小板计数1 579×109/L,腹部多普勒超声提示脾肿大;患者平素无疲倦乏力及纳差,无早饱感,无夜间盗汗、消瘦,无左上腹疼痛,无头晕、头痛,无畏寒、发热,无牙龈出血及皮肤瘀点、瘀斑,无黑便、血便,无胸闷、气促、心悸等不适。当地医院建议其至上级医院就诊,患者遂来我院就诊,急诊复查血常规示白细胞计数121.95×109/L,血红蛋白130 g/L,血小板计数1 504×109/L,以“白细胞及血小板增多”收入院。患者既往身体健康,无高血压、糖尿病、心脏病病史,无肝炎、结核病史,无放射性及毒物接触史,无特殊不良嗜好,家族中无类似疾病病史。入院体格检查:神志清楚,无贫血貌,皮肤无瘀点、瘀斑,浅表淋巴结未触及肿大,胸骨无压痛,心肺无异常,腹软,无压痛、反跳痛及肌紧张,肝脏肋下未扪及,脾脏肋缘下约2 cm,无摩擦感。入院后再次复查血常规示白细胞计数122.31×109/L,血红蛋白127 g/L,血小板计数1 469×109/L;白细胞手工分类示中性粒细胞0.40,淋巴细胞0.06,单核细胞0.04,嗜酸性粒细胞0.11,嗜碱性粒细胞0.12,幼稚细胞0.27。肝肾功能、血脂正常,乳酸脱氢酶445 U/L(正常值范围100~350 U/L),碱性磷酸酶79 U/L(正常值范围45~125 U/L),凝血常规无明显异常。腹部多普勒超声提示脾大。骨髓涂片示有核细胞增生活跃;粒系比例增高占骨髓有核细胞 (ANC)0.83,以中性粒细胞及以下阶段增生为主,原始粒细胞占ANC 0.005,嗜碱性粒细胞比例偏高,嗜酸性粒细胞比例增高;红系增生减少,形态未见明显异常,成熟红细胞形态及染色体大致正常;淋巴细胞比例降低,形态大致正常;单核细胞形态及比例大致正常;全片见巨核细胞39个,可见体积偏大的巨核细胞,散在血小板易见,成堆及成簇血小板增多;全片未见寄生虫;中性粒细胞碱性磷酸酶(NAP)积分17分。 骨髓活组织检查(简称活检):骨髓增生极度活跃(造血容量约95%),粒红系比例增高,可见典型不成熟前体细胞异常定位(ALIP)现象,幼稚细胞稍增多,小簇状或散在分布;粒系增生,各阶段细胞均可见,以偏成熟阶段细胞为主,嗜酸性粒细胞增多,小簇状或散在分布;可见少量中晚幼红细胞;巨核细胞数量增多,大部分胞体偏小,胞核分叶少;淋巴细胞、浆细胞散在分布可见;骨髓间质局灶可见纤维化,骨小梁规整。先后2次骨髓穿刺,定量聚合酶链反应(PCR)检测BCR-ABL1融合基因(p210型、p190型、p230型)均呈阴性。骨髓JAK2基因V617F突变、CALR基因突变、MPL基因突变阴性。骨髓染色体核型分析(细胞培养法G显带):46,XY,t(4;22)(p16;q11.2),del(7)(p13p21) [20](图1)。荧光原位杂交(fluorescence in situ hybridization,FISH)技术检测到BCR-ABL1融合基因。分析200个细胞,各信号模式如下:1F2R1G 54.5%,1F1R1G 33.0%,1F2R2G 6.5%,1F1R2G 2.0%,2R2G 4%(图2)。
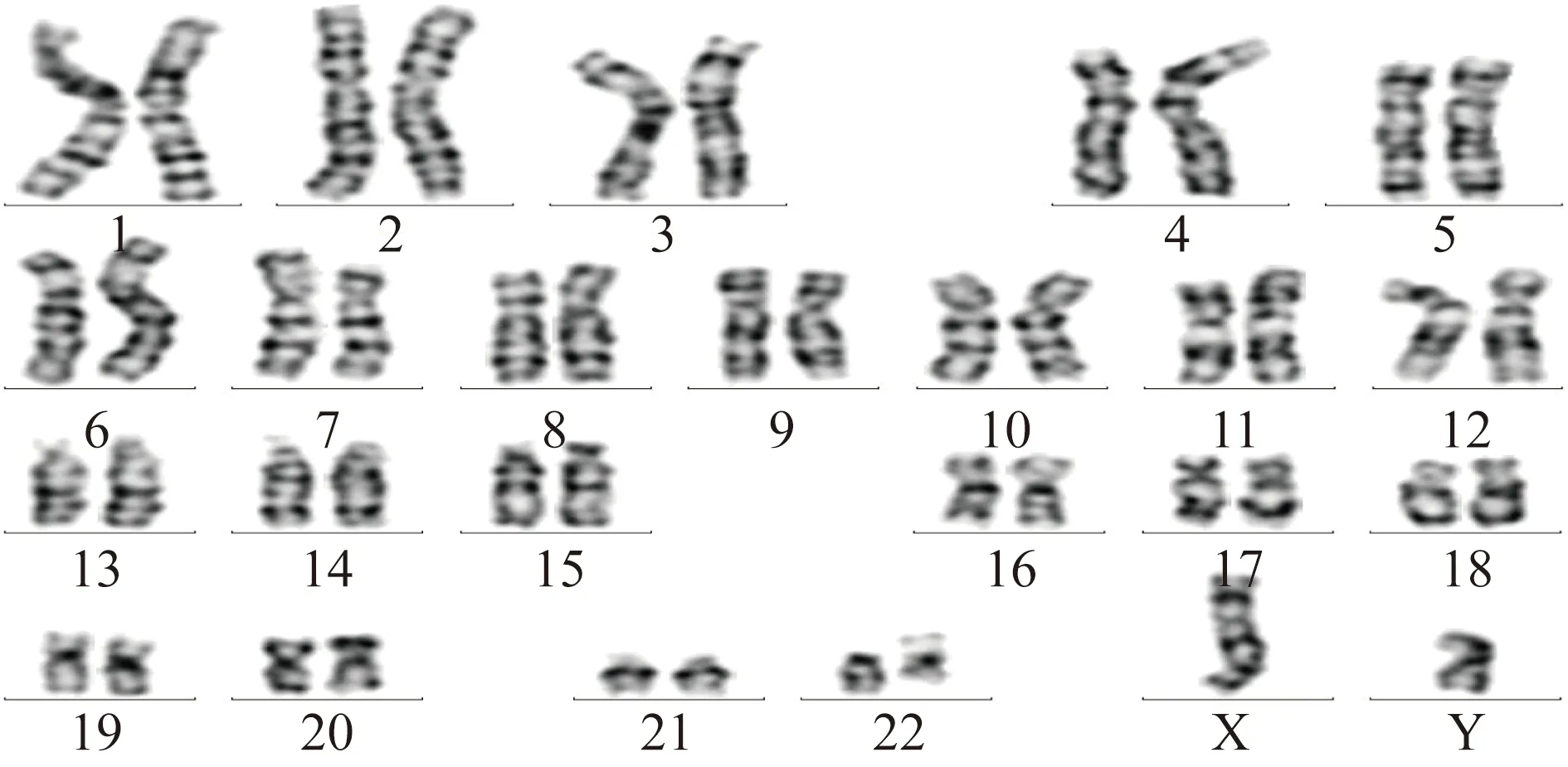
图1 细胞培养法G显带骨髓染色体核型分析结果
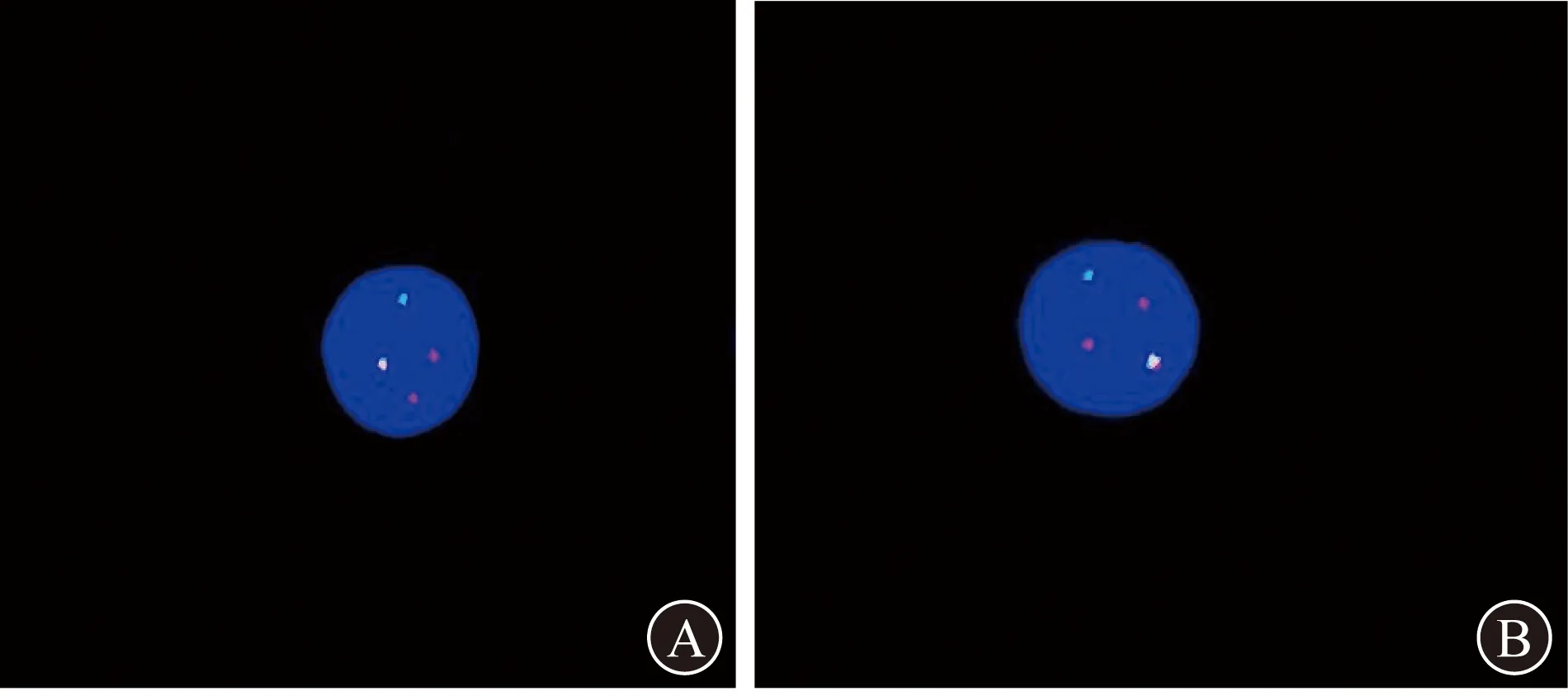
A、B 1F2R1G信号模式,绿色荧光G标记BCR(22q11)探针,红色荧光R标记ABL1(9q34)探针,BCR-ABL1融合基因显示黄色或红绿叠加信号F,正常信号模式2R2G,经典Ph染色体信号模式2F1R1G。阈值标准:融合信号1F>11%为阳性,2F>2%为阳性
结合临床表现及实验室检查,慢性粒细胞白血病(CML)慢性期诊断明确。入院后予羟基脲1~3 g/d治疗,根据血常规结果调整其用量,辅以碱化尿液、补液等处理。于2020年12月29日复查血常规示白细胞计数19.38×109/L,血红蛋白120 g/L,血小板计数1 522×109/L。患者为求进一步治疗至上级医院,予以甲磺酸氟马替尼片600 mg/d、注射用重组人干扰素α1b 50 μg/d治疗,于2021年1月21日复查血常规示白细胞计数7.45×109/L,血红蛋白125 g/L,血小板计数386×109/L。出院后患者长期服用甲磺酸氟马替尼片600 mg/d单药治疗,定期复查血常规均无明显异常,目前正在密切随访中。
2 讨 论CML是骨髓造血干细胞克隆增生的恶性肿瘤,通常以外周血白细胞计数显著升高为特点,其中以中、晚幼粒细胞为主,同时伴有嗜酸性粒细胞和嗜碱性粒细胞比例升高。大约有50%患者在初诊时血小板计数升高,部分患者可达1 000×109/L以上,且增高的程度与白细胞计数无明显相关性。持续性血小板计数>1 000×109/L为CML加速期的诊断标准之一,而在CML慢性期少见[1]。研究[2]结果表明,以血小板计数显著升高为表现的CML,在骨髓中可见到较多的不分叶圆核小巨核细胞。CML与原发性血小板增多症(ET)均属骨髓增殖性肿瘤,WHO的ET诊断标准中必须先排除BCR-ABL1融合基因或Ph染色体阳性[3]。ET的主要表现是外周血血小板计数显著升高并且存在功能异常,骨髓涂片通常显示以巨核细胞增生为主,粒系和红系增生正常,巨核细胞呈松散簇,大成熟巨核细胞增多,并且多存在JAK2 V617F、CALR、MPL基因突变[4]。本例患者血常规提示血小板计数显著升高,需与ET进行鉴别,但该患者外周血白细胞计数显著升高且外周血及骨髓中嗜酸性粒细胞、嗜碱性粒细胞增多,超声提示脾肿大,骨髓检查提示粒系增生活跃,巨核细胞数量增多,大部分胞体偏小,胞核分叶少,并且JAK2 V617F、CALR、MPL基因突变均为阴性,FISH技术检测到BCR-ABL1融合基因,因此诊断CML明确。
目前CML中发现的BCR-ABL1融合基因主要有3种[5]。其中,M-BCR可形成2种BCR-ABL1融合转录本,即e13a2(b2a2)和e14a2(b3a2),蛋白质产物均为P210;m-BCR可形成e1a2融合转录本,编码P190融合蛋白;u-BCR形成e19a2融合转录本,翻译成P230蛋白质产物。本例患者骨髓BCR-ABL1融合基因(p210型、p190型、p230型)阴性,但通过FISH技术检测到BCR-ABL1融合基因。究其原因,一方面存在定量PCR检测BCR-ABL1融合基因出现假阴性可能,但2次结果均出现假阴性的可能性小;另一方面很可能存在少见型BCR-ABL1融合基因转录本。据报道,目前有十余种少见型BCR-ABL1融合基因转录本,如b2a3(e13a3)[6]、b3a3(e14a3)[7]、e1a3[8]、e6a2[9]、e8a2[10]、e18-int-a2等,其中BCR基因断裂点可发生在外显子内部或内含子的插入,ABL1基因融合位点为外显子a3处[11-12]。在少见型BCR-ABL1融合基因转录本中又以b2a3、b3a3稍多见,其他则相对少见,并且b2a3、b3a3转录本CML患者的疾病进展相对缓慢,对酪氨酸激酶抑制剂(TKI)类药物敏感[6-7],而e6a2、e8a2转录本患者疾病进展较快且对TKI类药物耐药多见[10,13]。目前看来,本例CML患者对TKI类药物的治疗反应尚可。RNA测序又称转录组高通量测序,检测细胞内所有表达的转录本,可发现新的融合基因及位点,同时可行表达分析、可变剪切分析及突变分析等。针对BCR-ABL1融合基因的靶向RNA测序灵敏度更高,可检出低丰度的融合基因。本例患者可进一步行RNA测序检测出罕见型BCR-ABL1融合基因及位点甚至其他融合基因,但患者及其家属拒绝行该项检查。
95%以上初诊CML患者的染色体核型分析发现Ph染色体,即t(9,22)(q34,q11);极少数存在隐匿性Ph染色体,同时合并其他染色体异常。约有10%初诊CML患者存在变异性Ph染色体,可以由22q11与非9号染色体易位形成,也可以由9、22号及其他染色体复杂易位形成,无论是经典还是复杂易位的Ph染色体,9q34和22q11融合是其形成的根本。FISH技术是目前探究变异性Ph染色体产生机制的重要手段之一。变异性Ph染色体的形成主要为一步机制和二步机制[14],其中一步机制为染色体易位同时发生在3条及以上不同染色体之间(图3A),二步机制是在标准Ph染色体的基础上涉及另一染色体的相继易位(图3B)。大约有15%的经典易位和40%的变异易位通过FISH技术检测发现存在衍生9号和22号染色体BCR和(或)ABL1区域的缺失[14-15]。研究[14]结果表明,不论变异性Ph染色体为何种形成机制、参与染色体的数目或者存在BCR和(或)ABL1区域缺失与否,其对TKI类药物的分子生物学和细胞遗传学反应、疗效均无显著差异。本例患者骨髓染色体核型分析显示20个中期分裂相均存在4p16和22q11.2相互易位,FISH技术检测骨髓BCR-ABL1融合基因阳性,可能存在4、9、22号染色体三联易位。考虑可能由于Ph染色体易位拷贝数少或者9号染色体在易位前后长度未发生变化,常规染色体核型分析因不如FISH技术检测灵敏度高而不能得出阳性结果。根据相关文献研究结合该患者FISH技术检测信号模式,推测该患者染色体三联易位可能由于二步机制合并衍生9号染色体BCR和(或)ABL1区域的缺失所致(图3C和图3D)。

A 为一步机制(1F2R2G:1F位于衍生22号染色体,2R位于正常和衍生9号染色体,2G位于正常22号染色体和衍生另一染色体) B 为二步机制(2F1R1G:2F位于衍生22号染色体和另一染色体,1R位于正常9号染色体,1G位于正常22号染色体) C 为本例患者可能的染色体易位机制一(1F2R1G:存在衍生9号染色体BCR区域缺失,1F位于衍生22号染色体,2R位于正常9号染色体和衍生4号染色体,1G位于正常22号染色体) D 为本例患者可能的染色体易位机制二(1F1R1G:存在衍生9号染色体BCR和ABL1区域缺失,1F位于衍生22号染色体,1R位于正常9号染色体,1G位于正常22号染色体)
此外,推测该患者可能存在更为复杂的易位机制[14-15],同样可通过RNA测序予以证实。
在临床工作中遇到白细胞及血小板计数显著升高的患者必须常规进行BCR-ABL1融合基因、JAK2基因V617F突变,以及CALR、MPL基因突变检测,以排除其他骨髓增殖性疾病,若PCR检测BCR-ABL1融合基因阴性,但临床表现及骨髓细胞学提示CML可能,需进一步行BCR-ABL1融合基因FISH技术检测甚至RNA测序检查。
猜你喜欢
中国卫生标准管理(2022年21期)2023-01-03
健康体检与管理(2022年2期)2022-04-15
医学研究杂志(2021年10期)2021-11-26
中老年保健(2021年8期)2021-08-24
智慧健康(2021年33期)2021-03-16
医学概论(2020年46期)2020-11-22
医学新知(2019年4期)2020-01-02
文苑(2018年18期)2018-11-08
诗林(2016年5期)2016-10-25
医学研究杂志(2015年2期)2015-06-10
