
CDAN1基因突变导致的胎儿期非免疫性水肿的家系遗传学分析
2022-01-12 03:33王燕超孙筱放李少英何健淳张敏聪黄玲玲何文智
南方医科大学学报 2021年12期
王燕超,黎 青,孙筱放,李少英,何健淳,张敏聪,黄玲玲,何文智
1广州医科大学附属第三医院妇产科研究所实验部,广东 广州 510150;2广东省产科重大疾病重点实验室,广东 广州 510150;3广东省普通高校生殖与遗传重点实验室,广东 广州510150
非免疫性水肿(NIHF)是目前引起胎儿水肿的最常见原因,研究表明遗传因素(如单基因遗传病、染色体结构或数目异常)、血液病、感染和心血管疾病均可导致非免疫性胎儿水肿[1-3]。先天性红细胞生成障碍性贫血(CDA)是一种罕见的孟德尔遗传病,该病以大红细胞贫血、无效红细胞生成和发育不良为主要特征[4]。OMIM数据库根据临床症状和突变基因的不同,可将CDA分为5种类型[5]:即CDA I型(CDAN1基因和C15ORF41基因)、CDA Ⅱ型(SEC23B基因)、CDA Ⅲ型(KIF23基因)、CDA Ⅳ型(KLF1基因)和XLTDA(GATA1基因)。约75%CDA I 型是由于15号常染色体上CDAN1基因复合杂合突变所致[6],其特点是在儿童、青少年或以后的生活中出现中度至重度的大细胞性贫血[7]。目前,国内暂无CDA I型基因检测相关中文研究报道,国际上有关该疾病的研究对象大多为青少年和儿童,且主要侧重研究患者血液学参数与红细胞形态变化以及分析该病的临床症状和治疗方案。有关该疾病在胎儿期的报道非常少,研究者发现该基因变异可导致胎儿溶血性贫血、水肿、肝脾肿大和严重肺动脉高压[6,8]。总而言之,该基因不同位点的变异可表现在不同时期(胎儿期、儿童期、青少年期和成人期)和不同严重程度(从中度至重度贫血)。分析该疾病在各个不同时期的临床表现,有助于扩展临床医生对该病的病程认识,加深对基因型-表现型关联的理解,也有助于临床对胎儿水肿的病因学认识。
本研究采用全外显子组测序技术(WES)分析了1例先天性红细胞生成障碍性贫血1型家系,检测到引产胎儿的CDAN1基因存在复合杂合突变,找到了该家系孕期胎儿非免疫性水肿的遗传学病因,为该家系后续生育指导和出生缺陷防控提供了科学解释和理论依据。
1 资料和方法
1.1 研究对象
孕妇22周岁,G1P1,于我院产前诊断科就诊,孕17周超声提示胎儿水肿综合征,肝脾增大,心包缺损,孕妇本人要求终止妊娠,并行羊膜腔引产术查因。本研究经广州医科大学附属第三医院医学伦理委员会批准(医伦会审[2021]第115号),所有基因诊断均取得患者家属的同意,并签署知情同意书。
1.2 方法
1.2.1 样本DNA提取 采集胎儿引产组织和羊水细胞及其父母外周静脉血各5 mL,EDTA-K2 抗凝。采用Qiagen 微量DNA 提取试剂盒(Qiagen),按照说明书提取基因组DNA,并放置-20 ℃保存备用。
1.2.2 羊水细胞培养和G显带核型分析 取20 mL羊水分2 管,转速1800 r/min 离心10 min,弃上清,留取约0.5 mL细胞悬液,加入1 mL新鲜羊水培养基,混匀,37 ℃,5%CO2进行原代培养,收获细胞后按照标准流程进行G显带染色,按照中华人民共和国卫生行业标准(WS322.2-2010)分析羊水细胞染色体,常规计数20个细胞,分析3~5个核型。
1.2.3 全基因组拷贝数变异测序 采用基因组拷贝数异常检测试剂盒(贝康医疗)进行文库构建和后续实验,测序采用DA8600(达安基因)高通量测序仪对引产组织样本100 kb以上已知明确致病的CNⅤs,以及1 Mb以上的片段缺失/重复进行检测。
1.2.4 全外显子组测序 使用Agilent's SureSelectXT Human All Exon Ⅴ6(Agilent)进行外显子捕获测序,使用Illumina NovaSeq 6000测序系统进行人类全基因外显子组高通量测序。
1.2.5 测序结果分析 采用BWA软件对所获得的序列进行比对,使用GATK 对序列进行重排,并对变异(SNPs、indels)进行识别、注释和统计分析。
1.2.6 变异位点检测 用Sanger测序对先证者及家系成员进行突变检测和验证。采用Primer Primer5.0软件设计CDAN1基因:c.1264和c.2140位点上下游序列扩增引物,由上海生工科技有限公司合成,引物序列为:CDAN1-F1:GGCTGCTGTGCTTCTCACC,CDAN1-R1:GTCATCTCATTCCCTCCCCA。CDAN1-F2:CT GGATGTGCGGACTCTGC,CDAN1-R2:TGGGGAC TGTCTCTCAGCG PCR扩增产物用ABI3500XL测序仪(美国Applied Biosystems公司)进行一代测序。使用DNASTAR软件查看Sanger测序结果并与人类基因组序列进行比较。
1.2.7 变异位点的致病性分析CNⅤ-Seq 将测序得到的reads与已知人类参考基因组(hg19)进行比对。CNⅤ片段结果与DECIPHER、OMIM、DGⅤ、C1inⅤar 等数据库进行比对和关联表型的注释。WES-Trios:查询HGMD(Human Gene Mutation Database)、SNP/Ex AC、gnomAD和1000 Genomes Project数据库,对新发现的变异位点采用SIFT、I-mutant2、PolyPhen-2 及PROⅤEAN生物信息学软件进行蛋白功能预测。同时根据美国医学遗传学与基因组学学会(ACMG)2015年发布的基因变异解读标准和指南进行变异致病性分析。
1.2.8 变异位点的氨基酸保守性分析 并用Clustal Omega软件分析突变位点蛋白质序列的保守性。
1.2.9 变异蛋白空间结构模型分析 针对变异位点用Alpha Fold 2(https://alphafold.ebi.ac.uk)对CDAN1 的结构进行建模,预测错义突变前后对蛋白质空间结构的改变,采用PyMOL 2.3software对蛋白质的三维结构进行可视化分析。
2 结果
2.1 先证者主要临床特征
临床超声提示:全身皮下广泛水肿,胸腹腔积液,肝脾增大,胎盘增厚,心包缺损,股骨、头围<-2SD。引产后胎儿组织可见胎儿水肿(图1A)其他相关检查:孕妇22周岁,Rh(D)血型阳性,不规则抗体阴性,血常规结果正常,胎儿腹水常规正常,排除常见免疫性贫血所致胎儿水肿的可能。胎儿父母非近亲结婚且表型正常,孕期无用药史或不良环境接触史。
2.2 羊水细胞培养和G显带核型分析
46,XY,未发现异常核型。
2.3 全基因组拷贝数变异测序
检测范围内未见明显异常。
2.4 全外显子组测序数据质量
全外显子组双向测序目标捕获区平均测序深度为163.51×,测序目标区域覆盖度为99.90%,目标区域捕获均一性为为93.85%,覆盖30×以上区域占比为94.40%,Q30为94.40%。设计质量符合实验设计要求。
2.5 基因检测结果
测序数据经dbSNP、Ex AC和1000 Genomes数据库的过滤筛选后,发现了胎儿组织15 号染色体上的CDAN1基因(NM_138477.4)存在两个突变,经父母数据比对确认为复合杂合突变。包括8号外显子上的移码突变c.1264_1265delCT(p.Leu422Glyfs*16),和14号外显子上的错义突变c.2140C>T(p.Arg714Trp)(图1B,表1)。

表1 先证者家系CDAN1基因突变位点Tab.1 Mutation site of CDAN1 gene in the proband's family
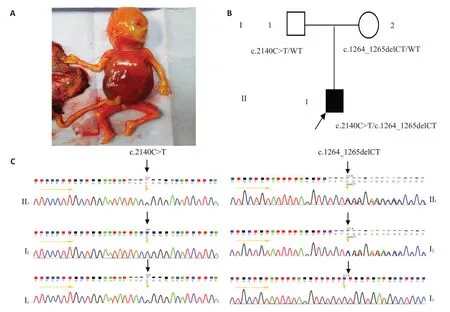
图1 家系CDAN1基因结果Fig.1 Gene sequencing result of the proband's family.A:Abnormal subcutaneous edema,ascites,pleural effusion of the affected individuals.B:Pedigree of the family's CDAN1 variations in the proband's family.C:Sanger sequencing revealed that the variation occurred at the c.2140 and c.1264_1265 sites in CDAN1 gene.The heterozygous mutations of the family at positions c.2140C>T and c.1264_1265delCT.
2.6 变异位点的致病性分析
CDAN1 基因14 号外显子上的杂合错义突变c.2140C>T,可导致其编码的第714 位氨基酸精氨酸(Arg)被色氨酸(Trp)取代,Clustal Omega软件分析该位点编码的第714位氨基酸在人类、鼠、长臂猿、猴、牛、猪、马、犬、大熊猫的物种进化过程中均高度保守(图2A)。PolyPhen-2、I-Mutant v2.0、Mutation Taster 和PROⅤEAN等生物信息学预测其编码蛋白有害或降解(表2)。蛋白模型分析显示p.Arg714Trp变异后,取代后的氨基酸之间的氢键和疏水作用力改变,致使蛋白质空间结构发生改变(图2B)。该变异在ClinⅤar数据库中记录为可能致病性,在HGMD数据库中记录为致病性。在ExAC数据库正常对照人群频率为0.0000417,在gnomAD 数据库正常对照人群中频率为0.00007569。有文献报道[1,9-11],在先天性红细胞生成障碍性贫血1型患者中检测到该变异与其他可能致病性或临床意义未明变异构成复合杂合突变结合已知致病性证据,根据ACMG遗传变异分类标准,判断该变异为可能致病性(PM2+PM3+PS3_Supporting+PP3)。
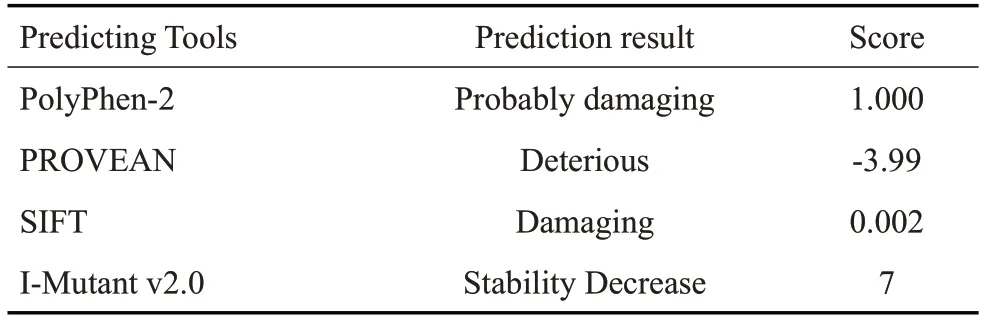
表2 生物信息学预测CDAN1基因c.2140C>T位点的致病性Tab.2 In silico analysis for predicting the pathogenicity of site c.2140C>T
CDAN1 基因8 号外显子上的杂合移码突变c.1264_1265delCT,导致蛋白编码在第422 位亮氨酸(Leu)由甘氨酸(Gly)取代,并在随后15位提前出现氨基酸的终止密码,变异截短大于10%的蛋白质,可能发生NMD(Nonsense-mediated mRNA decay)导致蛋白功能受损,AutoPⅤS1等生物信息学预测其编码蛋白有害。蛋白模型分析显示变异后,形成了截短的CDAN1蛋白质(图2C)。该变异在ClinⅤar数据库和HGMD数据库中暂无记录,在ExAC数据库正常对照人群频率为0.000008237,在gnomAD数据库正常对照人群频率为0.000003976,根据ACMG遗传变异分类标准,判断该变异为可能致病性(PⅤS1+PM2)。
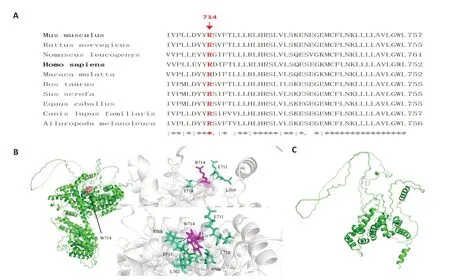
图2 生物信息学预测和蛋白3D结构可视化Fig.2 Bioinformatics and three-dimensional protein structure visualization.A:Conservative analysis of amino acid site 714 of Codanin-1 protein.B:The structure of Codanin-1 is modeled to predict the effect of missense mutations on protein structure using Alpha Fold 2.Arrow on the left shows the location of amino acid site 714,and the hydrogen bonds and hydrophobic forces between the amino acids and changes in protein spatial structure are shown on the right.Blue dotted lines are hydrogen bond interactions,and yellow dotted lines are hydrophobic interactions.C:Prediction diagram of truncated Codanin-1 protein model.
2.7 Sanger测序验证分析
对该家系成员3人CDAN1基因进行Sanger测序验证,先证者(Ⅱ1)c.2140C>T/c.1264_1265delCT 复合杂合突变,其父(I1)为c.2140C>T/WT杂合突变,其母(I2)为c.1264_1265delCT杂合突变,与WES结果一致(图1C)。
3 讨论
胎儿水肿(HF)发病率是1/1700到1/3000,当胎儿有至少两个不同部位的液体积聚时,可诊断为胎儿水肿[12]。随着抗Rh(D)免疫球蛋白的临床应用,免疫性水肿的比例已明显下降,非免疫性水肿(NIHF)所占比例逐渐升高。然而,由于NIHF的病因复杂且临床预后较差,大部分致病原因和致病机制仍不清楚,其发病率、死亡率和复发风险因病因的不同而有很大差异。因此,对遗传学病因的确定,有益于临床医生对疾病的预后判断、复发风险评估和遗传咨询[2,13,14]。我们的研究结果说明,当NIHF排除染色体非整倍体和拷贝数变异的因素后,全外显子测序技术将是一种可行且有效的分子诊断方法。
先天性红细胞生成障碍性贫血(CDA)是一种非常罕见的遗传病,该病的特征为成红细胞形态异常,红细胞无效生成,大细胞贫血,部分患者可表现为骨骼发育异常、指甲发育不全和脊柱侧弯。由于统计样本量、种族和基因诊断能力的差异,目前该疾病的总人群发病率暂不清楚。目前,未见基于中国人群的发病率统计和系统性研究[15]。CDA的临床症状异质性比较大,从无症状者到重度贫血均可出现。有些亚型,如CDAⅡ型容易被误诊为其他类型的贫血或由于国家发展程度和机构检测能力的差异而未被发现。值得指出的是,虽然通过光学显微镜和骨髓穿刺等传统技术手段可以诊断CDA,但却不能将CDA的亚型进行很好的区分,因此全外显子测序可作为一种创伤小而操作性强的替代方法[5,16]。
CDAN1基因的28个外显子位于染色体15q15,跨越基因组1.5 kb,编码含有1227个氨基酸的codanin-1蛋白,该蛋白是一种O-糖基化蛋白,可能与核膜完整性、微管附着有关[11,17]。CDAN1基因突变最常见于新生儿和青少年。在新生儿期临床表现为肝脏肿大、早期黄疸和宫内生长迟缓,在青少年期,大多数受累者终身伴有中度至重度贫血,通常伴有黄疸、脾肿大和胆结石,部分存在脊柱侧凸和四肢畸形[1,6,8,17-19]。仅有极少数病例报道了妊娠期的胎儿水肿,且除两篇报道外,大多研究报道均未行相关基因检测,仅根据外周血象和骨髓穿刺结果进行了临床确诊[20-23]。2019年新加坡的一篇研究发现了一例患胎在孕19周被诊断为胎儿水肿,表现为肝脾肿大、腹水、胎盘增厚、四肢发育落后和心包积液伴心脏扩大[9],随后,该研究者检测出CDAN1基因c.1744C>Tp.Q582*和c.2140C>T复合杂合。该胎儿在宫内多次输血治疗,最终在出生后5个月死于多器官功能衰竭和脓毒血症。上述病例与本研究的患胎均携带了同样的c.2140C>T错义突变,和突变位点不同的移码突变——前者为c.1744C>T,本例为c.1264_1265delCT,为国内外首次报道的突变,可能因产生了截短蛋白而影响蛋白质功能,因此,在临床症状上有相似的表现——均在中孕期发生了明显的胎儿水肿等一系列表型。这提示了我们可根据突变类型和突变位点对患儿愈后作出一定程度的预判。
Liu等[20]报道了一例中国患胎在孕22周B超发现胎儿水肿、腹水伴心脏扩大,胎儿大脑中动脉收缩期峰值流速(MCA-PSⅤ)升高,脐血穿刺检测提示胎儿严重贫血(Hb 6.5 g/dL),WES检测出CDAN1基因c.2059C>T(p.R687C)纯合突变,最终孕妇决定终止妊娠。该病例与本研究病例表现型相似,且均未发现胎儿四肢结构异常,本研究病例由于胎儿孕周较小脐带细,未能抽取脐血行相关血液学检测。
目前,已在多名CDA 患者中检出c.2140C>T(R714W)突变,研究表明该区域位于CpG岛上,被认为是CDAN1的突变热点区域[1,17,24],且目前已报道的致病性突变多集中在12-24号外显子上,提示该区域突变可能导致更严重的临床症状[24]。
然而,CDAN1基因突变类型和临床严重程度的相关性仍不清楚,本研究暂时无法解释部分儿童患者脊柱侧凸、四肢畸形和指甲发育不全的原因和机制,且多数报道由于当时检测方法所限,均未行全外显子组测序,或仅检测了CDAN1基因,是否存在其他共效基因影响患者临床表型,有待进一步研究。
综上,通过家系全外显子组测序方法,我们对一例不明原因的胎儿期非免疫性水肿家系进行遗传学分析,发现了CDAN1基因c.2140C>T/c.1264_1265delCT复合杂合突变。本研究首次报道了CDAN1基因8号外显子的一个新的突变,丰富了CDA致病基因的突变列表,及其在妊娠期胎儿水肿综合征中发挥的作用,同时加深了我们对该基因表现型和个体表现度差异性的认识,为该家系的遗传咨询、生育指导提供相应理论支持。
猜你喜欢
安徽农业大学学报(2022年2期)2022-11-09
电子科技大学学报(2022年5期)2022-10-29
中国土壤与肥料(2021年5期)2021-12-02
南京医科大学学报(自然科学版)(2021年8期)2021-10-19
种子(2021年3期)2021-04-12
中国生殖健康(2020年4期)2021-01-18
校园英语·下旬(2017年7期)2017-07-14
科技视界(2016年27期)2017-03-14
中学生理科应试(2016年7期)2016-05-14
中国动物保健(2015年4期)2015-10-21
