
基于密码子优化策略的无乳链球菌表面蛋白LrrG的原核表达、纯化及免疫原性
2022-01-10 01:44陈徵婷可小丽陈刚黄丽芳刘志刚卢迈新
大连海洋大学学报 2021年6期
陈徵婷,可小丽,陈刚,黄丽芳,刘志刚,卢迈新
(1.中国水产科学研究院珠江水产研究所 农业农村部热带亚热带水产资源利用与养殖重点试验室,广东省水产动物免疫技术重点实验室,广东 广州 510380; 2.广东海洋大学 水产学院,广东 湛江 524025; 3.仲恺农业工程学院 动物科技学院,广东 广州 510550)
无乳链球菌Streptococcusagalactiae隶属于链球菌属Streptococcus,依据Lancefield氏血清分类法将其归为B族,故无乳链球菌也称B族链球菌,简称GBS,该菌是导致人畜共患病的重要细菌性病原之一[1-2]。1966年,Robinson等[3]在淡水鱼金色美鳊Notemigonuscrysoleucas中首次分离到GBS。此后,多种海水和淡水鱼类均出现感染GBS的报道,如鲱Brevoortiapatronus、条纹胭脂鱼Mugilcephalus、大西洋黄鱼Micropogoniasundulatus、银鳟Cynoscionnothus等海水鱼[4-5],易感淡水鱼包括胭脂鱼Lizaklunzingeri、斑马鱼Daniorerio、金头鲷Sparusauratus、银鲳Pampusargenteus、石斑鱼Epinepheluslanceolatus、宝石鲈Scortumbarcoo、虹鳟Oncorhynchusmykiss等[6-8],淡水罗非鱼Oreochromisniloticus是近10年来受无乳链球菌感染最严重的宿主[9-12]。目前,有效防治仍然是链球菌病研究的重点,在水产养殖生产过程中,虽然抗生素是一种比较快捷实用的方法,但其存在严重的耐药风险[13-14],而疫苗作为免疫防治的重要手段之一,具有高效、特异、无污染等优点,是水产病害防控的良好选择。
目前,鱼类无乳链球菌疫苗大部分仍处于研发阶段,如灭活疫苗[15]、减毒疫苗[12,16]及各类基因工程疫苗等[17-19],其中,基因工程疫苗的关键是获得优质的候选抗原。无乳链球菌荚膜多糖在毒力方面起着重要作用,且具强有效的抗原性,因此,基于其荚膜多糖相关疫苗一直是研究热点,然而,荚膜多糖基因具有多态性,导致其跨血清型交叉免疫保护一直未能获得理想的效果。研究表明,GBS表面蛋白抗原作为疫苗候选抗原或特异性GBS多糖的载体蛋白[20],具有大多数GBS血清型中保守的抗原靶点,是跨血清保护性疫苗的良好候选者,LrrG蛋白便是其中之一。
LrrG是无乳链球菌表面蛋白之一,LrrG基因编码一种新的富含亮氨酸的重复序列(LRR)基序,与细菌侵袭素共同存在。重组蛋白LrrG具有GBS黏附素的功能,在小鼠体内具有较高的免疫原性,可诱导特异性IgG,相对免疫保护率达91%[21],且其编码的新型富亮氨酸重复序列(LRR)在各无乳链球菌血清型中(Ⅰa、Ⅰb、Ⅱ~Ⅷ)均十分保守[22]。本研究团队前期曾利用原核表达的LrrG重组蛋白作为疫苗抗原对罗非鱼无乳链球菌病进行免疫保护试验,口服或注射免疫条件下均显示具有良好的免疫原性,相对免疫保护率达69%~77%[23],不足是LrrG可溶性上清重组蛋白的表达量有限。
密码子优化是通过在信使RNA编码区进行密码子同义突变以增加蛋白表达产量的重要方法,目前在生物制药领域应用十分广泛[24]。基于前期研究基础,本研究中尝试通过密码子优化获得LrrG重组蛋白的可溶性上清蛋白,同时对优化后的重组蛋白进行免疫原性验证,以期为无乳链球菌LrrG表面蛋白基因工程疫苗的制备提供科学数据。
1 材料与方法
1.1 材料
试剂:胶回收、质粒提取试剂盒购自Tiangen公司;pCzn1质粒、BL21(Plys)表达菌株由南京钟鼎生物技术有限公司提供;限制性内切酶、Pfu DNA 聚合酶购自TaKaRa公司;Protein Marker购自Thermo公司;T4 DNA连接酶购自 Promega公司;IPTG、Acr、Bis、Tris购自Sigma 公司;SDS购自Amresco公司;TEMED购自BIO-RAD公司;Tyrptone、Yeast Extract购自Oxoid公司;0.22 μm无菌过滤器、透析袋购自Millipore公司;DNA胶纯化试剂盒购自Axygen公司;SDS-PAGE凝胶配制、BCA蛋白检测试剂盒购自上海碧云天生物技术有限公司;Binding buffer、Elution buffer购自生工生物工程(上海)股份有限公司;His Bind亲和纯化试剂盒购自BIO Basic公司;PVDF膜购自Pall公司;鼠抗His单克隆抗体购自Abmart公司;辣根过氧化物酶(HRP)标记的羊抗鼠抗体购自武汉博士德生物工程有限公司;ECL化学发光液购自Millipore公司;TMB 显色液购自Biopanda公司;其他试剂均为国产分析纯或化学纯。
仪器:Mini Protean Ⅱ垂直平板电泳系统、Gel Doc2000成像系统、水平电泳系统(均为美国BIO-RAD 公司);PTC-200基因扩增仪(美国MJ Research公司);320-S pH 计(美国 Mettler Toledo公司);AR5120电子天平(美国Aohaus公司);MultiTemp III恒温水浴锅、Hofer UV-25紫外透射仪(美国Amersham Pharmacia 公司);雪花状制冰机(日本Sanyo公司);JY92-2D 超声波细胞粉碎机(中国新芝科器研究所);NANODROP2000(Thermo公司)。
1.2 方法
1.2.1LrrG基因优化与合成 根据本实验室分离并保存的罗非鱼源无乳链球菌(Ⅰa,ST7)的LrrG基因序列(登录号: KC920814.1),利用密码子优化软件(南京钟鼎生物技术有限公司)对LrrG基因进行优化,在不改变氨基酸序列的前提下,利用宿主细胞对密码子的偏好性,将低频稀有密码子替换为使用频率较高的密码子,同时对编码疏水区和信号肽区的基因序列进行分析,并在优化后基因引物的两端分别加入NdeⅠ、XbaⅠ酶切位点和 His 标签序列。根据优化后的基因序列,采用PAS (PCR-based Accurate Synthesis)的方法合成基因,设计全长拼接引物,引物为Primer-F: ACGCCATATCGCCGAAAGG;Primer-R: GGCAGGGATCTTAGATTCTG。PCR扩增产物经琼脂糖凝胶电泳检测后,用DNA凝胶回收试剂盒回收目的片段。
1.2.2 pCzn1-LrrG重组表达载体的构建 利用双酶切法,将LrrG基因插入到表达载体pCzn1的NdeⅠ和XbaⅠ间。首先将pCzn1载体用NdeⅠ和XbaⅠ酶进行双酶切,然后再与LrrG基因连接,酶切体系为:pCzn1载体5.0 μL,10×FD buffer 5.0 μL,NdeⅠ (10 U/μL)2.5 μL,XbaⅠ(10 U/μL)2.5 μL,用ddH2O补足至50.0 μL,37 ℃下酶切1 h。酶切产物经琼脂糖凝胶电泳检测后,用胶回收试剂盒回收,将回收的酶切产物和目的DNA片段用连接酶在37 ℃下连接25 min。连接体系为:LrrGDNA 4.0 μL,线性载体pCzn1 3.5 μL,T4连接酶(5 U/μL)2.5 μL。
1.2.3 pCzn1-LrrG重组载体的转化、筛选与鉴定
将连接产物pCzn1-LrrG转入大肠杆菌感受态中,向感受态细胞中加入连接液10 μL,于冰上放置30 min。将样品管迅速置于42 ℃水浴中热激90 s,之后迅速置于冰上冷却5 min,再向管中加入800 μL LB液体培养基。于37 ℃下以220 r/min振荡培养50 min后,涂布于含Amp的筛选平板上,正面向上37 ℃下静置1 h,待菌液完全被培养基吸收后倒置培养皿,37 ℃下静置培养16~20 h。次日观察菌落,挑取单菌落接种于液体LB培养基,提取质粒,进行双酶切鉴定。酶切体系为:质粒3 μL,NdeⅠ和XbaⅠ酶各0.25 μL,10×buffer 1.0 μL,ddH2O 6.5 μL。经菌液PCR和双酶切后,进行测序(广州艾基生物有限公司)。
1.2.4 表达菌株的制备 将质粒1 μL加入100 μL大肠杆菌表达菌株BL21(Plys)感受态细胞中,置于冰上20 min,42 ℃下热激90 s,迅速置于冰中5 min,加入600 μL LB培养液,37 ℃下以200 r/min振荡培养1 h后,涂布于含50 μg/mL Amp的LB平板上,37 ℃下倒置培养过夜。次日挑取单菌落接种到含有50 μg/mL氨苄霉素的LB培养基中,37 ℃下振荡培养3 h,取0.5 mL菌液与0.5 mL 50%的无菌甘油混合,-80 ℃下保存备用。
1.2.5 优化后LrrG重组蛋白的诱导表达 挑取转化好的表达菌株单克隆,接种于含50 μg/mL AMP的4 mL LB培养液试管中,于37 ℃下以220 r/min振摇过夜,次日按1∶100接种于含50 μg/mL AMP的200 mL LB培养液中,37 ℃下以220 r/min振摇至菌体OD600 nm值为0.6,取出1 mL培养物,以10 000g室温离心2 min,弃上清,用100 μL Binding buffer缓冲液重悬菌体沉淀,向剩余的培养物中加入IPTG至终浓度为0.5 mmol/L,37 ℃下以220 r/min振摇6 h,诱导融合蛋白表达,取出1 mL培养物,室温下以10 000g离心2 min,弃上清,用100 μL Binding buffer重悬菌体沉淀。剩余培养物以4 000g离心10 min,弃上清,用Binding buffer重悬菌体沉淀,重悬液用冰浴超声波破碎60 min,超声时间5 s,间隔10 s,功率300 W,然后分别取上清液与沉淀液加入上样缓冲液重悬。用质量分数为12% 的SDS-PAGE凝胶进行电泳分析,考马斯亮蓝染色检测。
1.2.6 优化后LrrG重组蛋白的纯化及与优化前LrrG重组蛋白表达量的比较 优化前LrrG重组蛋白利用PET-32a-LrrG重组表达菌株,依照陈雪[23]所述表达条件进行,并确保其诱导表达前的OD值与优化后LrrG重组蛋白pCzn1-LrrG的OD值保持一致。利用NI-IDA纯化树脂预装柱子进行纯化,采用重力法使存储缓冲液完全流出。加入2倍柱体积的Binding buffer 到预装柱中进行平衡,控制流速约为1 mL/min。将蛋白和Binding buffer混匀,使其总体积相当于两个柱体积。将混合液加入柱子,收集流穿液到离心管中。用两倍柱体积的Binding buffer清洗柱子并收集流穿液。用两倍柱体积的Elution Buffer洗脱柱上的蛋白,此步骤重复两次。将洗脱的蛋白进行SDS-PAGE分析,并采用ImageJ软件分析优化前后LrrG重组蛋白上清表达量的相对差异。
1.2.7 优化后LrrG重组蛋白的Western blot鉴定
SDS-PAGE电泳完毕后,将蛋白条带转印至聚偏二氟乙烯膜(PVDF膜)上,转印后PVDF膜用质量分数5%的脱脂奶粉室温封闭30 min,分别采用两套抗体进行Western blot鉴定:用一抗鼠抗His-tag(兔抗GBS多克隆抗体)(1∶5 000)和二抗HRP标记的羊抗鼠(HRP标记的羊抗兔)(1∶5 000),先后与转印后的PVDF膜反应,用显影、定影试剂进行显影和定影,观察结果。
1.2.8 优化后LrrG重组蛋白的免疫原性 选取同等大小规格的健康吉福罗非鱼(体质量为3.4 g±0.8 g,体长为4.2 cm±0.2 cm),试验前已暂养2周,将试验鱼随机分为4组(每组50尾),LrrG重组蛋白免疫组和对照组(PBS代替蛋白)各2组,试验组免疫剂量为2 μg/g(蛋白质量/鱼体质量),用PBS将优化后的LrrG可溶重组蛋白稀释至所需浓度,与弗氏不完全佐剂1∶1混合后,进行腹腔注射免疫,对照组注射等体积的PBS。同时,利用相同规格的健康吉福罗非鱼进行无乳链球菌毒株(LZ1F-Ia-ST7)的半致死剂量LD50检测,具体参考陈雪[23]的方法。免疫两周后,利用LZ1F-Ia-ST7 LD50对所有免疫鱼体进行腹腔注射,连续记录死亡情况,2周后统计各组死亡率。
相对免疫保护率(relative percent survival,RPS)计算公式为
RPS=(1-试验组死亡率/对照组死亡率)×100%。
2 结果与分析
2.1 罗非鱼源无乳链球菌LrrG基因的优化
经分析发现,LrrG基因序列中存在6个原核表达体系中使用频率低于20%的稀有密码子,决定对其进行替换优化。其次,蛋白N端第763AA~775AA为较强的疏水区,经预测N端759AA~778AA为典型的跨膜结构,无信号肽结构,因此,决定去掉N端第763 AA之后的序列进行优化表达。原始LrrG基因序列全长为2 361 bp,优化后的基因加入了酶切位点、His标签等,去掉了无信号肽的疏水区基因序列。经测序,优化后的完整LrrG基因序列全长为2 286 bp,G+C含量有明显变化,优化前后分别为34.94%和45.67%,符合G+C含量在30%~70%为理想区间的标准。氨基酸序列完全一致,优化前后基因序列比对结果如图1所示。利用软件对密码子优化后的LrrG基因预测的LrrG蛋白氨基酸序列进行分析,分子结构式为C3862H6182N1084O1162S12。预测的蛋白理论相对分子质量为86 777,理论等电点pI值为9.17,不稳定性指数为30.53,因大于阈值40时性质不稳定,说明优化后的蛋白性质较为稳定。

Origina为原始LrrG基因序列;Optimized为优化后的LrrG基因序列;下划线为优化后基因与原始基因的差异碱基位点;实线方框内为起始密码子;虚线方框内为终止密码子。Origina represents the original sequence of the LrrG gene; Optimized represents the optimized sequence of the LrrG gene; The differential nucleotide sites between the original and optimized sequence of the LrrG gene are shown by underlines; Initiation codon is shown in the solid box and the stop codon is shown in the dotted box.图1 LrrG 基因优化前后核苷酸序列对比Fig.1 Alignment of nucleotide sequences between the original LrrG gene and the optimized LrrG gene
2.2 重组质粒pCzn1-LrrG的构建与鉴定
将优化的目的基因片段和线性化的pCzn1载体进行连接(图2),连接液转化至大肠杆菌感受态细胞,构建无乳链球菌表面蛋白LrrG的重组表达载体pCzn1-LrrG。将重组质粒进行双酶切,结果显示,重组质粒电泳后呈现环状、线性和超螺旋3种基本形态,酶切后的pCzn1线性载体和LrrG基因片段大小与理论值(4 400 bp和2 286 bp)相符,且酶切条带单一,表明重组载体构建成功(图3)。利用引物(pCzn1-F: ACGCCATATCGCCGAAAGG;pCzn1-R: GGCAGGGATCTTAGATTCTG)进行测序鉴定,结果显示,插入片段碱基序列与优化后的LrrG基因碱基序列一致,其编码的氨基酸序列与优化前编码的氨基酸序列完全一致,表明LrrG优化基因插入片段完全正确,未发生碱基突变。
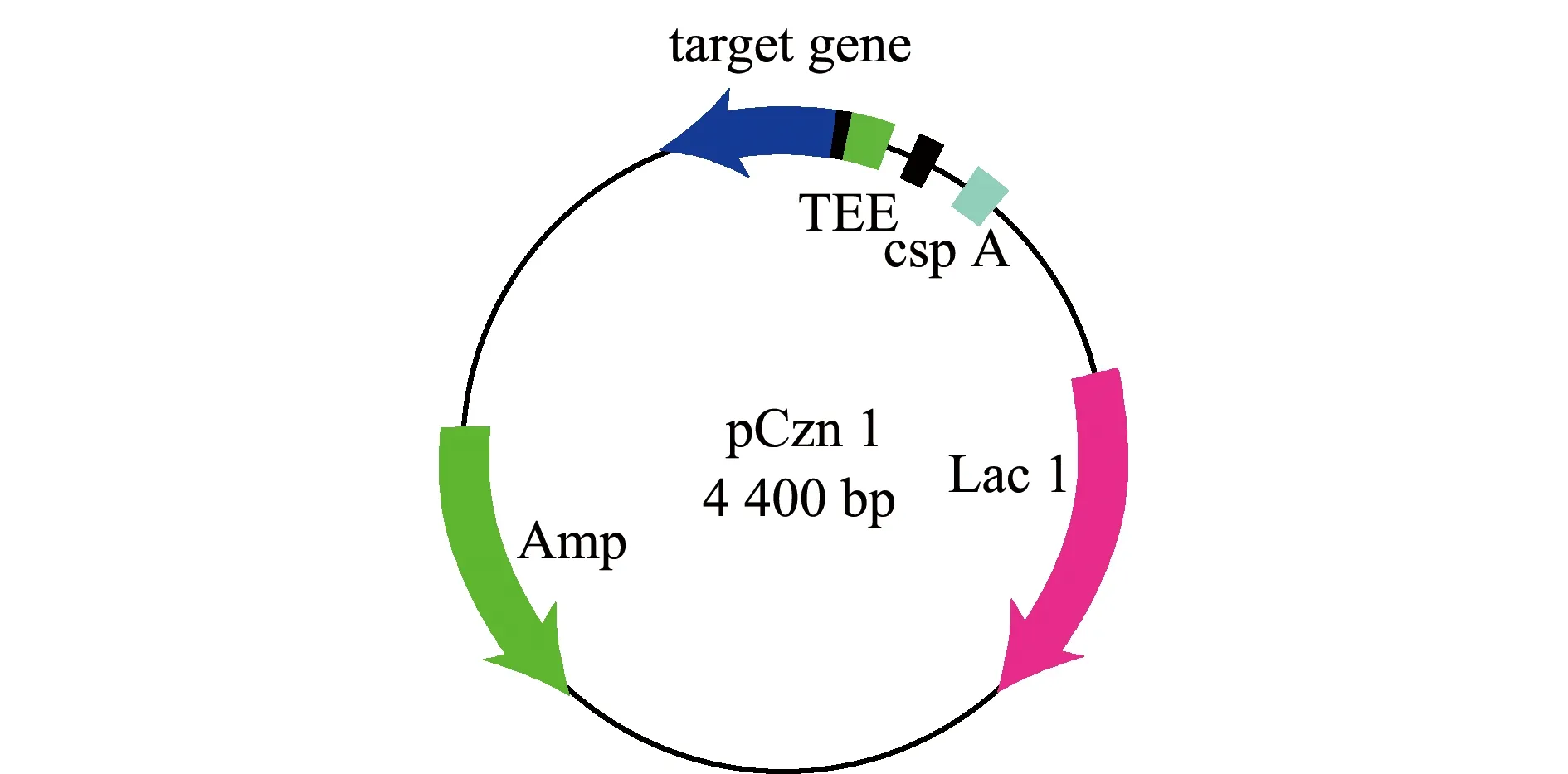
图2 pCzn1载体构建图谱Fig.2 pCzn1 vector construction map

1—酶切前质粒;2—酶切后质粒;M—DL4500 DNA marker。1—plasmid before restriction digestion; 2—plasmid after restriction digestion; M—DL4500 DNA marker.图3 重组质粒pCzn1-LrrG的酶切Fig.3 Restriction digestion of recombinant plasmid pCzn1-LrrG
2.3 优化后LrrG重组蛋白的诱导表达
测序验证正确的pCzn1-LrrG表达菌株单克隆扩大培养后,利用IPTG诱导蛋白表达,在37 ℃下诱导培养6 h,以未加入IPTG作为对照。经12% SDS-PAGE凝胶电泳分析,目标蛋白主要存在于上清中,这说明,37 ℃诱导条件下LrrG蛋白在原核表达宿主BL21(Plys)中已成功诱导表达,该蛋白相对分子质量为108 000(图4)。

M—蛋白分子量标准;1—未诱导总蛋白;2—诱导总蛋白;3—诱导上清;4—诱导包涵体。M—protein marker; 1—uninduced total protein; 2—induced total protein; 3—induced supernatant; 4—induced inclusion bodies.图4 优化后His-LrrG重组蛋白的SDS-PAGEFig.4 SDS-PAGE of codon optimized His-LrrG recombinant protein
2.4 LrrG重组蛋白的纯化及其优化前后蛋白表达量的比较
诱导表达后的菌液经超声波破碎后,对上清液进行Ni-IDA 柱亲和层析纯化,随后利用SDS-PAGE对纯化的蛋白进行检测。SDS-PAGE分析结果(图5)显示,菌体沉淀溶解后的上清液在相对分子质量为108 000附近出现高浓度的目的条带,且条带单一。不同批次诱导表达后,上清稳定表达,且优化后LrrG重组蛋白的表达量为1.5~1.8 mg/mL,优化前LrrG重组蛋白的表达量为0.43~0.74 mg/mL(图5、图6)。用ImageJ软件分析累积光密度显示,优化后的表达量相对优化前提高了2.2~3.8倍。

M—蛋白分子量标准; 1—未诱导; 2—流过液; 3—洗脱后的可溶蛋白。M—protein marker; 1—uninduced; 2—liquid flowed through column; 3—eluted soluble protein.图5 优化后His-LrrG重组蛋白纯化的SDS-PAGEFig.5 SDS-PAGE of codon optimized His-LrrG fusion protein purification

M—蛋白分子量标准;1—未诱导重组菌全菌蛋白;2—重组菌诱导产物;3—诱导上清;4—洗脱后的可溶蛋白。M—protein marker; 1—the uninduced recombinant total bacterial protein; 2—the induced recombinant total bacterial protein; 3—the induced supernatant; 4—eluted soluble protein.图6 优化前His-LrrG重组蛋白纯化的SDS-PAGEFig.6 SDS-PAGE of His-LrrG fusion protein purification
2.5 优化后LrrG重组蛋白的Western blot鉴定
在Western blot试验中,重组蛋白LrrG与一抗为鼠抗His-tag单克隆抗体和兔抗GBS多克隆抗体均可以发生反应,在相对分子质量约108 000附近均有特异杂交条带,与SDS-PAGE结果一致,表明优化后的重组蛋白LrrG具有GBS抗原反应原性(图7)。

1—一抗为鼠抗His-tag单克隆抗体;2—一抗为兔抗GBS多克隆抗体。1—primary antibody is mouse anti His-tag monoclonal antibody; 2—primary antibody is rabbit anti GBS polyclonal antibody.图7 重组蛋白LrrG的Western blot鉴定Fig.7 Western blot identification of recombinant protein LrrG
2.6 优化后LrrG重组蛋白的免疫原性
用优化后的可溶蛋白LrrG免疫罗非鱼2周后,利用无乳链球菌毒株LZ1F-Ia-ST7半致死剂量1.19×106CFU/mL进行人工攻毒LrrG免疫组和对照组罗非鱼。结果显示:蛋白免疫组死亡率为16%~20%,平均死亡率为18%,相对免疫保护率为61.54%~69.23%,平均相对免疫保护率为65.39%;PBS对照组死亡率为50%~54%,平均死亡率为52%(表1)。
3 讨论
3.1 罗非鱼源无乳链球菌重组LrrG蛋白的优化表达
罗非鱼链球菌病传染性强、死亡率高,给罗非鱼产业造成了严重的经济损失[25-26]。其主要病原无乳链球菌,根据其荚膜多糖(CPS)基因座序列的不同,可分为Ⅰa、Ⅰb、Ⅱ、Ⅲ、Ⅳ、Ⅴ、Ⅵ、Ⅶ、Ⅷ、Ⅸ等10个血清型[27],虽然目前Ⅰa(92.3%)和Ⅰb(7.7%)是罗非鱼无乳链球菌病的两种主要血清型[28],但后期菌株变异的风险也不可忽视。
目前,基于无乳链球菌保守表面蛋白的新疫苗候选蛋白已开展大量研究,包括LrrG、ScpB、Lmb、表面免疫原性蛋白Sip,以及含有C蛋白α和β成分的肋骨和串联重复序列等[29]。其中,LrrG蛋白编码一种新的LPXTG锚定的表面抗原,该表面抗原含有细菌入侵素和LRR蛋白家族等成员中发现的富亮氨酸重复序列(LRR)受体[30]。重组LrrG蛋白在体外显示出以依赖的方式黏附于上皮细胞,表明它可能在GBS中起黏附因子的作用,且在小鼠和罗非鱼中均表现出较好的相对免疫保护率[22-23]。为进一步提高重组LrrG蛋白在大肠杆菌原核表达的上清表达量,本研究中对其进行了密码子优化。大肠杆菌是体外重组蛋白生产的优选宿主,因为其生长快,易于操作,培养成本低,且遗传学研究充分。但是在异源蛋白表达过程中,因异源基因可能只包含少数可在表达宿主中使用的密码子,原始的编码序列在新的宿主中通常会阻止或限制重组蛋白的表达,因此,重组蛋白通常较难在其原始环境之外表达[31]。大肠杆菌也不例外,利用大肠杆菌进行重组蛋白的表达,在大肠杆菌中稀有密码子的存在通常会对蛋白表达产生影响,容易造成重组蛋白的异源表达量低下[32]。一般情况下,在大肠杆菌中生产可溶性蛋白比总蛋白表达困难得多,通常需要优化宿主菌株和培养条件,以及筛选最佳的基因片段和融合标签[33-35]。所以,可以通过密码子优化的策略来解决异源表达基因的问题,进而提高蛋白表达量[36]。本研究中,通过替换LrrG蛋白中大肠杆菌稀有密码子,并进一步优化其疏水区和无信号肽区域等,重新构建了高表达载体pCzn1-LrrG,获得了大肠杆菌BL21(Plys)重组表达菌株。研究发现,优化后的重组菌株LrrG上清表达量明显高于优化前,表明按照大肠杆菌密码子进行优化可以提高外源原核生物基因编码的蛋白在大肠杆菌中的表达量,这与其他研究者的结论相符。何明娟等[37]通过密码子优化的办法在大肠杆菌中高效表达了马槟榔甜蛋白。李文锋等[38]通过密码子优化提高了新型鸭呼肠孤病毒σB蛋白原核表达量。高见等[39]优化了高危型人乳头瘤病毒L2E7基因密码子,Wang等[40]优化了人类膀抑素C基因密码子,均使目标蛋白的原核表达量得到了不同程度的提高。其中,潘群兴等[41]优化鸡传染性法氏囊病病毒VP2基因密码子,结果显示,密码子优化型重组菌表达量能提升至野生型重组融合蛋白GST-VP2表达量的2倍。本研究中,通过比较优化前后的LrrG重组蛋白的表达量,发现优化后LrrG重组蛋白的上清表达量较优化前提高了2.2~3.8倍。
3.2 优化后LrrG重组蛋白的免疫原性
优化后的蛋白抗原是否保持其原有的免疫原性,是决定其是否可用于后续应用的关键。本研究中,首先通过Western blot鉴定发现,优化后的LrrG重组蛋白不仅可以和鼠抗His-tag发生特异性结合,也可以和兔抗GBS多克隆抗体发生特异性结合,说明密码子优化后的LrrG重组蛋白具有GBS抗原反应原性。进一步通过LrrG注射免疫罗非鱼,发现优化后的LrrG重组蛋白对罗非鱼抗GBS感染具有61.54%~69.23%的相对免疫保护率,平均约为65.39%,这与优化前的相对免疫保护性结果相似[23],说明优化后的LrrG重组蛋白仍然具有较好的免疫原性。该结果为无乳链球菌表面蛋白疫苗的研发及应用提供了科学参考。
4 结论
1) 重组表达载体pCzn1可用于优化后LrrG重组蛋白的表达。
2) 通过密码子优化可提高无乳链球菌表面蛋白LrrG在大肠杆菌中的表达量。
3) 优化后的LrrG重组蛋白仍然具有较好的免疫原性。
猜你喜欢
渔业致富指南(2022年4期)2022-11-05
广西植物(2022年8期)2022-09-07
当代水产(2022年6期)2022-06-29
家庭医学(2021年11期)2021-12-29
今日农业(2021年15期)2021-11-26
皮肤病与性病(2021年3期)2021-07-30
发明与创新·中学生(2019年6期)2019-06-26
农民致富之友(2019年7期)2019-05-23
湖北农业科学(2018年15期)2018-11-12
生物学教学(2018年2期)2018-08-07
