
新型铈掺杂氧化铋复合催化材料制备及其光催化性能研究
2022-01-07 14:09包海峰欧阳文璟严雨阳郑雨莹
吉林化工学院学报 2021年11期
包海峰,欧阳文璟,严雨阳,郑雨莹
(吉林化工学院 资源与环境工程学院,吉林 吉林 132022)
近年来,因半导体光催化氧化技术具有的独特优势,在难降解污染物控制与降解领域中备受瞩目,Bi2O3属于P型半导体材料,许多研究表明Bi2O3在可见光范围内有良好的吸收,展现出了一定的可见光催化性能[1-2].同时研究也发现,单一Bi2O3在光催化反应过程中产生的电子-空穴对易再次重组复合,表现出较低的光量子产率,限制了其进一步的研究与应用.因此,采用技术手段将某金属掺杂进Bi2O3,研究制备一种新型复合光催化剂,加宽其可见光吸收带,并使之具有较小的能隙禁带宽度,以增强其可见光响应效率,提高光催化性能,便成为在光催化领域推广应用所必需的前期基础研究,具有重要的现实意义.稀土离子的电子层结构比较特殊,存在多电子组态,并且其光谱特性亦比较独特,许多研究表明可使用稀土元素对半导体进行特定的修饰改性,可以充当光生电子和空穴的捕获陷阱,更有利于光生电子-空穴的分离与利用,这已倍受研究者的青睐[3-4].在稀土金属中铈元素最丰富,利用金属Ce掺杂来提高一些半导体材料的可见光响应和催化性能已经有了部分报道[5-8].不同成分和形貌复合结构会表现出不同程度的光催化活性,在众多形貌中一维线性结构有着独特的传导特性和尺寸效应,本研究基于静电纺丝技术将Ce掺杂到Bi2O3中,对Bi2O3进行微观调控,研究制备性能增强的一维线性复合催化剂,增强复合材料的光催化效率,通过对等样品表征分析研究不同掺杂比例对改性后复合材料微观结构、尺寸形貌以及可见光响应性能的影响,并将所制备的催化剂应用于液相中可见光催化降解甲基橙染料有机污染物,以评价其性能,为Bi2O3的改性开发、新型复合光催化材料的研制及其在水处理领域光降解可生化性较差的有机污染物提供依据和支持.
1 实验部分
1.1 配制纺丝溶液
用二甲基甲酰胺(DMF)溶解一定量的Bi(NO3)3·5H2O,充分搅拌,形成溶液A;二甲基甲酰胺(DMF)溶解一定量的Ce(NO3)3·6H2O,充分搅拌,形成溶液B;用无水乙醇和二甲基甲酰胺(DMF)溶解一定量的聚乙烯基吡咯烷酮粉末(PVP,Mw=1 300 000),定量加入冰醋酸,充分搅拌,形成溶液C.控制不同的PVP wt%,取不同体积(mL)的A溶液和B溶液加入C溶液中,得到成分比例不同的前驱体溶液,静置陈化后待静电纺丝.
1.2 光催化材料制备
采用静电纺丝技术混纺所制备的前驱液,纺丝参数为收距15 cm,电12 kV,混纺得到Ce/Bi/PVP复合纤维,烘干后置于程序控温箱式炉内进行程序控温烧结,经退火后即得到Ce掺杂Bi2O3光催化剂样品.同时,本实验同步条件下制备纯Bi2O3样品和纯CeO2样品,用以对比分析.实验制备的光催化材料样品统一命名编号为CBE系列.复合材料制备实验流程如图1所示.
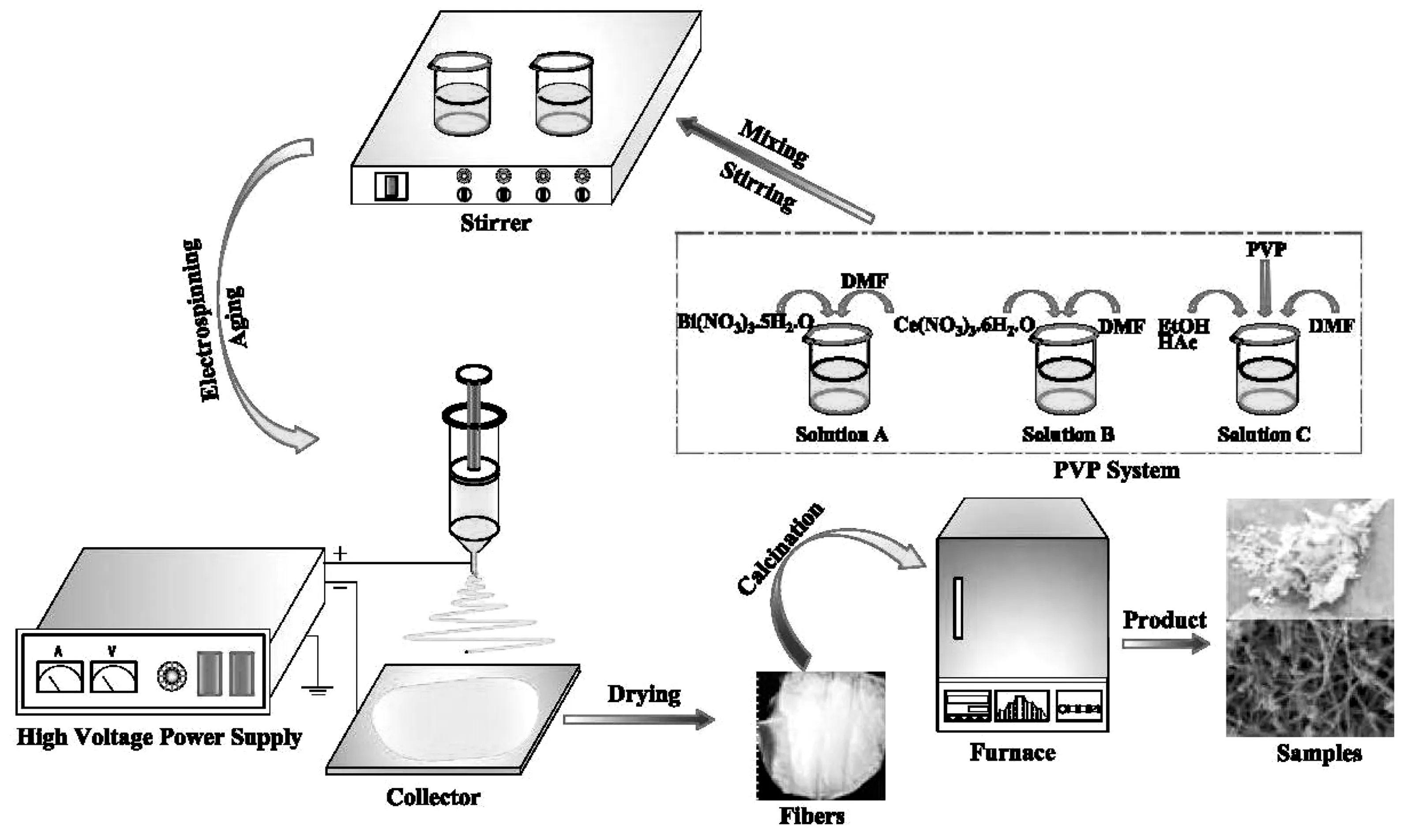
图1 光催化剂制备流程图
1.3 光催化材料催化性能的测试和评价
CBE催化剂用量为5.0 g/L,以甲基橙(MO)为目标污染物,浓度为10 mg/L,进行光催化降解染料有机污染物实验.整个催化体系先在黑暗中充分搅拌30 min后开启氙灯光源,进行光解反应.每隔15 min进行取样,水样经离心后取上清液检测吸光度值以及全谱线,分析对比吸光度值,检验不同铈掺杂比例条件催化剂对甲基橙(MO)的降解效率,评价CBE复合催化剂的性能.
2 结果与讨论
2.1 静电纺丝前驱液中高聚物PVP质量比(wt%)对纤维形貌和直径分布的影响
采用静电纺丝技术获得纤维时,前驱液中聚合物PVP wt%是影响前纺丝纤维形态及性质的重要因素,因此配制前驱液时调节聚合物和金属盐用量便可获得不同性质的纺丝溶胶溶液.本研究配制的前驱液中PVP wt%分别为5wt%、6wt%、7wt%、8wt%,用以确定催化材料制备时最佳的前驱液中高分子聚合物PVP的wt%.
图2为PVP wt%分别为5wt%~8wt%,陈化后在12 kV纺丝电压、1.0 mL/h纺丝液流速以及15 cm接收距离条件下混纺出的纤维SEM图.

(a)5wt%
由图2可见,不同PVP wt%条件下Bi/Ce/PVP纤维的形貌和纤维直径尺寸有所差异.当PVP wt%为5wt%时,纤维直径较细且伴有大小不一的珠状节点,见图2(a).增大PVP wt%,纤维上的珠状节点逐渐消失,PVP wt%在6wt%~8wt%范围时静电纺丝获得的纤维表面基本光滑,见图2(b)~(d),无珠状节点.由局部放大图可见,当PVP wt%从5wt%增大到8wt%时,随着PVP wt%的增大,线性形貌保持良好,但纤维直径由细逐渐增粗,从200 nm左右增粗到900 nm左右,这是由于纺丝过程参数一定条件下,随着前驱液PVP wt%增大,黏度也逐渐升高,但纤维直径过大不利于后续复合材料制备.
图3为PVP wt%为5wt%~8wt%的纺丝纤维直径分布统计.
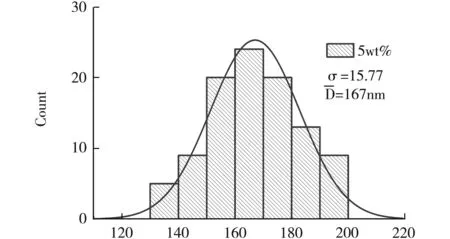
fiber diameter/nm(a)
采用Image-Pro Plus 6.0工具统计分析相应样品的100个分布数据,统计结果见表1.

表1 不同PVPwt%条件下纺丝纤维直径统计结果
表1结果结合图2表明,PVP wt%是静电纺丝纤维外观形貌和直径分布的重要影响因素.PVP质量占比较小时,前驱液黏度也较低,纤维表面有珠状节点存在,不利于后续材料制备;当PVP wt%为8wt%时,获得的纤维直径统计方差较大,形貌均匀性已经不理想;而在PVP wt%为7wt%条件下,获得的纺丝纤维形貌均匀性比较好,直径分布较优.从形貌的均一程度并综合考虑所获得的混纺纤维直径因素,确定PVP质量百分比为7wt%条件下静电纺丝纤维最适合后续材料的制备.
2.2 掺杂改性催化剂材料XRD图谱分析
图4为PVP wt%为7wt%条件下制备的Ce掺杂改性CBE系列光催化剂以及单纯CeO2样品和单纯Bi2O3样品的XRD图谱.

2 Theta/degree图4 Ce掺杂Bi2O3光催化剂的XRD图谱
由图4可见,纯Bi2O3(CBE1)样品对应于PDF#76-1730标准卡为单斜晶系的α-Bi2O3,其2θ主要特征衍射峰值在27.476°、33.347°处对应于(120)、(200)晶面.而纯CeO2(CBE2)样品则对应于PDF#65-5923标准卡,其2θ主要特征衍射峰值28.588°、47.555°和56.429°处则为(111)、(220)以及(311)晶面.经金属Ce掺后,在4个复合材料样品(CBE3、CBE4、CBE5、CBE6)的XRD谱图中均出现了Bi7.38Ce0.62O12.31相特征峰,对应于PDF#50-0372标准卡的四方晶系,这说明制备的Ce掺杂复合材料样品中存在有Bi、Ce的复合氧化物.在复合材料中,这些Bi、Ce复合氧化物可能是以固溶体的形式存在,或者均匀地分散于Bi2O3晶体中[9-10].但在所有复合催化剂样品XRD图谱中均没有出现Ce和Ce氧化物的特征衍射峰,说明Ce成功掺杂进了Bi2O3晶体中,并替换了部分Bi晶格位,图谱上表现出Bi2O3的特征衍射峰变弱变宽[11].和纯相Bi2O3和CeO2相比较,复合材料样品最强特征衍射峰位置介于两纯相之间,图谱上峰型基本对称,对应于PDF#78-1793标准卡属于四方晶系,其2θ峰值27.944°处为(201)晶面.综上表征分析说明,在复合样品中Ce被成功掺杂进去,Ce的掺杂使得单斜晶的Bi2O3晶相转变为复合材料的四方晶相[12].在所有Ce掺杂的复合材料样品图谱中只发现含有元素Bi、Ce、O、C,没有出现其他元素物质的特征衍射峰,表明复合材料样品纯净.此外,在单一的Bi2O3和所有复合材料的样品图谱中,存在次要相Bi2O2CO3的特征衍射峰,属于四方晶系,这与材料样品退火冷却制备时吸收一定量的CO2相关[13].
2.3 掺杂改性催化剂材料SEM图谱分析
半导体催化剂的形貌是影响其光催化性能的重要因素之一.图5为自制催化剂的SEM和能谱表征.

(a) CBE1
其中图5(a)~(f)为各CBE催化剂样品SEM图,图5(c)、(d)、(e)、(f)为复合材料SEM图,Ce掺杂比例(摩尔比)分别为0.05(CBE3)、0.25(CBE4)、0.5(CBE5)和0.75(CBE6),图5(a)、(b)则是相同方法制备的单一的纯Bi2O3(CBE1)和CeO2(CBE2)样品SEM图.
由图5可见,基于静电纺丝所制备的样品均显示出一维的线性结构.在图5(a)中(CBE1),呈现出Bi2O3颗粒晶体沿线性组装,图5(b)中(CBE2), CeO2片状晶体亦沿线性装配,两者均展现出了中空的管状结构,晶体呈现良好的生长状态,图谱上测量样品外径,CBE2(685 nm)比BCE1(228 nm)粗.当用Ce掺杂修饰后,见图5(c)~(f),不同掺杂量的复合材料仍为一维的线性结构,复合材料的直径尺寸随Ce掺杂比例由小至大表现出先缩小再增粗的趋势,但基本均在200 nm左右,对比发现0.25掺杂比例的CBE4样品直径最小,因此CBE4的比表面积更具有优势.同时,随着掺杂比例由0.05增大至0.75,初始50 nm左右壁厚的中空管状结构逐渐消失,表面外观形貌也逐渐变得密实、光滑,说明不同Ce掺杂比例对晶体的取向生长存在着不同的影响[14].SEM图中,在Ce掺杂后的复合材料表面均没有观察到CeO2片状结构晶体,这与复合材料XRD表征分析中没发现铈氧化物特征衍射峰的结果也相吻合,说明Ce掺杂后,在样品制备晶体生长过程中CeO2形成受到限制.图5(g)为CBE4复合纤维样品的SEM图,复合纤维为无序非晶态,且表面光滑,与图5(e)相比较可以看出,纤维直径(380 nm)经退火处理制备成复合催化剂后直径缩小(224 nm).图5(h)为CBE4的能谱图,图谱中有C、O、 Ce、Bi、Au元素,其中Au元素外来于检测时样品的表面镀金,与XRD物相表征分析相对照,表明所制备的CBE复合催化剂仅由C、O、 Ce、Bi元素构成,样品成分符合预期,成功制备了Ce掺杂Bi2O3的纯净复合催化剂材料.
2.4 掺杂改性催化剂材料TEM图谱分析
为了进一步获得复合催化剂微观形态结构的基本信息,选取0.25比例Ce掺杂CBE4样品进行TEM表征分析,其谱图如图6所示,进一步研究复合催化剂微观的形态结构.
在低倍镜图谱图6(a)中可以明显观察到复合材料为管状结构,直径约为200 nm左右,和SEM图谱基本相符,图中可见四角形状的微小晶粒按照线性取向装配,经估算这些微小的四角形状晶粒尺寸大小为15~20 nm,这与XRD表征分析相吻合.图6(b)(高分辨图)中可以看到样品结晶度良好,晶格纹络清晰可见,晶格间距测量结果表明为四方晶系的Bi2O3(201)晶面、(110)晶面、Bi7.38Ce0.62O12.31(400)晶面以及Bi2O2CO3(013),这与XRD表征分析相互佐证,表明成功地实现了Ce掺杂.在样品电子选区衍射图6(c)中,衍射环与衍射斑点共存,揭示出复合催化剂样品的多晶结构.

(a)低分辨
2.5 掺杂改性催化剂材料UV vis-DRS分析
光催化剂可见光响应范围是影响光催化反应的重要因素.图7为自制CBE光催化剂固体紫外-可见漫反射光谱(UV-vis DRS)图谱,图7内插图为Ce掺杂前后估算材料吸收边的对比图;经Kubelka-Munk方程估算的CBE催化剂禁带宽度结果如图8所示[15].

Wavelength/nm图7 CBE催化剂UV-vis DRS图谱
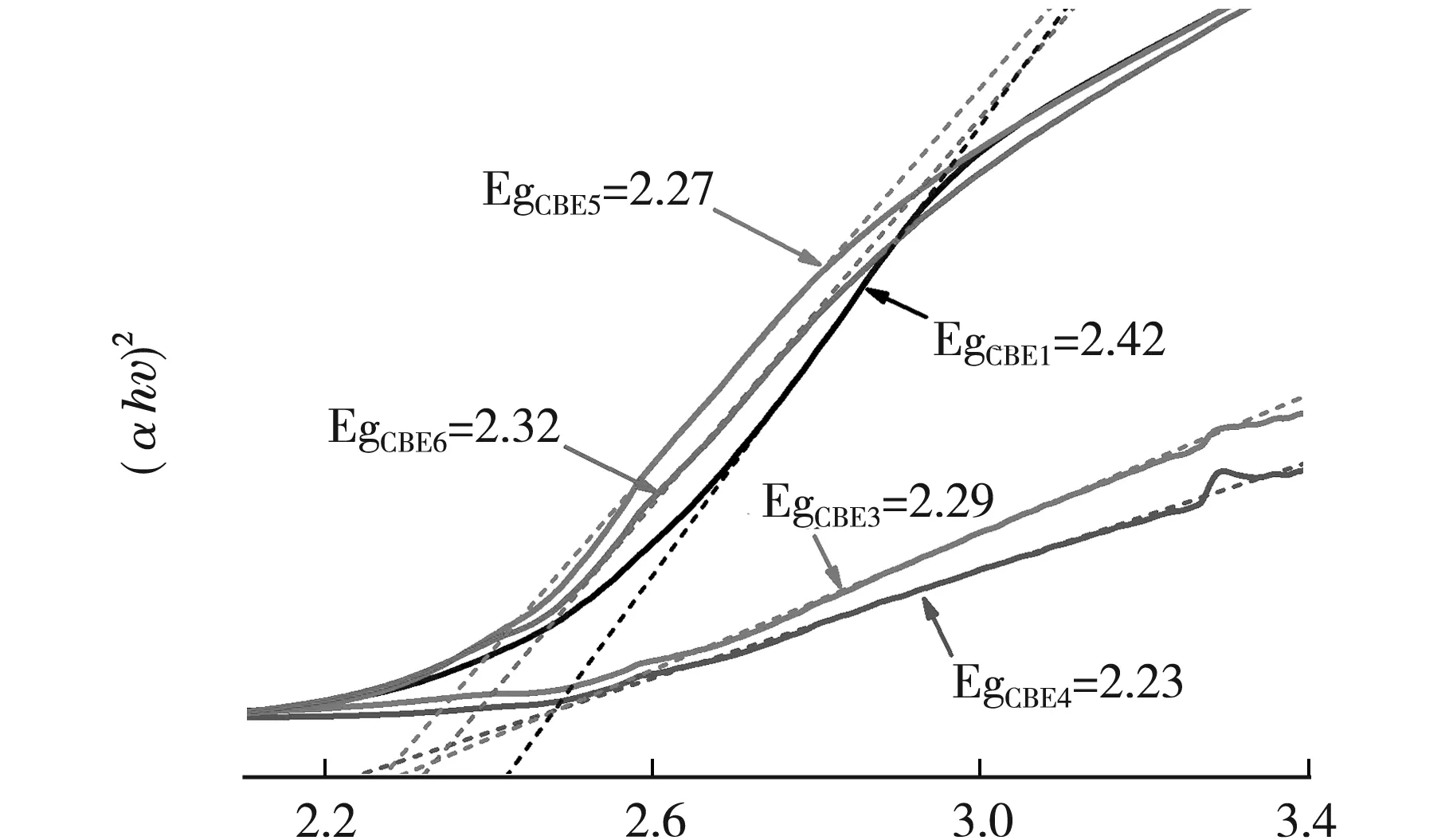
hv/eV图8 CBE催化剂禁带宽度估算
由图7可见,自制的BCE催化剂光吸收曲线呈陡峭线型,具有紫外-可见光响应效应,光吸收特性良好.陡峭型的吸收曲线也表明CBE催化剂的可见光响应是基于能级跃迁,而非杂质跃迁[16].内插图为CBE1(纯Bi2O3)和CBE4(0.25掺杂比例)样品的收边估算,吸收边分别为473 nm、558 nm,并同理求得CBE3、CBE5、CBE6的吸收边分别为539 nm、510 nm、506 nm.比较可知,经Ce掺杂的复合样品吸收边均发生了不同程度的红移,且均在可见光范围内,其中CBE4复合样品红移最大.根据Kubelka-Munk方程估算CBE各样品的禁带宽度Eg(图8),结果依次为2.62 eV(CBE1)、2.30 eV(CBE3)、2.22 eV(CBE4)、2.43 eV(CBE5)和2.45 eV(CBE6),对比可见,Ce掺杂后复合催化剂禁带宽度Eg值均缩小,其中0.25掺杂比例的复合催化剂(CBE4)禁带宽度最小.催化材料禁带宽度小则可见光吸收能力强,对可见光响应效果会更好,因此推测CBE4催化剂更有利于在可见光催化降解有机污染物.
2.6 掺杂改性催化剂材料催化活性分析与评价
以甲基橙(MO)为目标有机污染物,对所制备的CBE材料的可见光催化性能进行测试.图9为CBE催化剂光催化降解MO效果谱图.

t/min(a)光催化降解MO效率图
图9(a)中,没有催化剂时,光催化反应时间内体系MO浓度随时间进程几乎不变化;而存在CBE催化剂时, MO随着光照时间延长浓度逐渐降低,发生光降解.90 min光催化反应后,CBE1(纯Bi2O3)和CBE2(纯CeO2)MO的光降解率分别是36.06%、19.23%,相比较之下,经Ce掺杂修饰后的复合催化剂光催化均表现出较高催化性能,其中0.25比例Ce掺杂的CBE4催化剂具有最高的光催化降解MO的能力,90 min MO光降解率可达到98.81%,表明经Ce掺杂改性后,复合材料的催化性能得到了增强,这与前述表征分析结论也是相吻合的.从图9(b)中可以明显看到,MO吸收特征峰随光催化降解进程而逐渐降低,MO发生了光降解反应.利用Langmuir-Hinshelwood动力学模型对其光降解过程进行拟合[17],结果线性拟合关系良好,如图9(c)所示,各拟合的R2值均在0.95左右,说明CBE系列催化剂光催化降解MO过符合一级动力学方程.CBE各催化剂光降解动力学速率常数结果如图9(d)所示,其中CBE4的光降解速率常数(K值)最高,为0.062 02 min-1,是CBE1(纯Bi2O3)的8.7倍,表现出增强的催化活性.
3 结 论
基于静电纺丝技术成功制备出催化性能增强的一维线性管状金属Ce掺杂的Bi2O3新型复合催化剂.经表征、分析和光催化活性测试评价,结果表明:
(1)以聚乙烯基吡咯烷酮粉末(PVP,Mw=1 300 000)为静电纺丝前驱液中高分聚合物,在12 kV电压、15 cm接收距离、1 mL/h出流速度条件下混纺纤维,PVPwt%最优条件为7%,此时获得的纺丝纤维均匀性较好,直径分布和外观形貌较优, Bi/Ce/PVP混纺复合纤维直径统计分析为375±60 nm,有利于后续处理制备催化材料.

(3)金属Ce能以替换Bi晶格位的方式对Bi2O3进行掺杂改性,掺杂后会影响Bi2O3晶相形成,复合材料晶粒尺寸缩小.Ce掺杂后的复合材料吸收边产生红移,禁带宽度变小,可见光响应能力增强,复合材料光生电子-空穴对也因Ce的掺杂而提高了分离效率,光催化性能得以增强.
猜你喜欢
九江学院学报(自然科学版)(2022年2期)2022-07-02
云南化工(2021年7期)2021-12-21
少先队活动(2020年12期)2021-01-14
陶瓷学报(2019年5期)2019-01-12
中成药(2017年3期)2017-05-17
三峡大学学报(自然科学版)(2017年1期)2017-03-20
领导科学论坛(2016年9期)2016-06-05
中国资源综合利用(2016年9期)2016-01-22
燕山大学学报(2015年4期)2015-12-25
合成技术及应用(2015年3期)2015-12-11
