
CRISPRi/ddCpf1 促进大肠杆菌中丙二酰辅酶A 的积累
2022-01-07 08:54洪煦琳
食品与生物技术学报 2021年12期
洪煦琳, 贾 笑, 肖 毅
(上海交通大学生命科学技术学院,上海 200240)
丙二酰辅酶A(malonyl-CoA) 是食品、保健品等产品中多种有价值化合物合成的重要前体物质,比如次级代谢产物类黄酮、聚酮化合物和脂肪酸等[1-2]。 大多数宿主中,丙二酰辅酶A 通过乙酰辅酶A 羧化酶 (acetyl-CoA carboxylase,ACC) 催化乙酰辅酶A 羧化合成而来[3]。 由于许多次级代谢产物的天然生产菌株难以大规模培养、 基因操作困难,易遗传操作的大肠杆菌(Escherichia coli,E. coli)已成为次级代谢产物生产的常用微生物细胞工厂。 然而,E. coli 中的丙二酰辅酶A 被用于产生脂肪酸和磷脂,仅留下少量可用于其他次级代谢[4-5],是大规模生产目标次级代谢产物的潜在瓶颈[6]。 目前,通过基因调控提高丙二酰辅酶A 的含量,可以促进多种下游目标产物的生物合成,如白藜芦醇和柚皮苷等类黄酮化合物[7]。实验表明,丙二酰辅酶A 的常规基因敲除会明显影响细胞生长,导致生长迟缓甚至细胞死亡[8]。 而添加脂肪酸代谢抑制剂浅蓝菌素(cerulenin) 可以显著提高细胞内丙二酰辅酶A 的水平[9],这表明可以通过调控基因转录来提高丙二酰辅酶A 水平。
基于成簇的规律间隔的短回文重复序列(clustered regularly interspaced short palindromic repeats,CRISPR) 是一种适应性免疫系统, 常见于细菌和古细菌中,可以对外源侵入的核酸进行靶向切割,从而起到保护自身免受外源核酸侵害的免疫作用。 CRISPR 系统由Cas (CRISPR-associated protein) 蛋白和crRNA(CRISPR RNA) 相互作用形成核糖核蛋白复合体, 在成熟crRNA 的指导下,识别靶向DNA 序列并进行切割。 基因工程中,crRNA和tracrRNA 可以人工改造并融合成sgRNA(singlemolecule guide RNA) 用于基因组编辑[10]。 CRISPR干扰是一种基于CRISPR 系统的基因转录抑制技术, 利用催化失活的Cas 蛋白 (dead Cas protein,dCas)对特定基因转录进行下调[11]。以CRISPR/Cas9系统为例,dCas9 是Cas9 的催化失活突变体,dCas9-sgRNA 复合体能够与DNA 特异性结合而不进行核酸切割, 通过空间阻碍RNA 聚合酶(RNA polymerase,RNAP) 的转录过程,从而抑制目标基因的转录[11]。 代谢工程中,CRISPRi 技术可用于下调代谢竞争途径、抑制转运蛋白的活性等,从而提高目标产物的产量、减少毒性中间体和副产物[12]。目前,CRISPRi 已经被用于多种代谢工程宿主,包括Escherichia[13]、Mycobacterium[14]、Clostridium[15]、Corynebacterium[16]、Pseudomonas[17]。 Cpf1 是II 类Ⅴ-A 型CRISPR 系统, 具有和Cas9 同源的类RuvC 内切酶结构域[18]。 由于其具有组成简单、特异性高、能同时作用多靶点、脱靶效应更低[19]等优势,被视为常用基因编辑技术CRISPR-Cas9 的替代品,已经被开发成为多重基因编辑、基因调控的工具,应用于代谢工程[20-22]。 Ji 等用ddCpf1 替代化学抑制剂用于下调fabI 基因的表达,将丁烯酸的产量提高了6 倍[22]。Li 等在谷氨酸棒杆菌(Corynebacterium glutamicum)中应用CRISPR/dCpf1 抑制gltA、pck、pgi 和hom 基因(参与赖氨酸合成) 表达,将赖氨酸产量提高了4倍以上[23]。
噬菌体T7 的基因2 编码的一种大肠杆菌RNA聚合酶抑制剂Gp2 蛋白, 可以干扰DNA 在RNAP下游的正确定位,有效抑制E-σ70转录起始[24],表现了其在抑制基因转录中的调控潜力。 另外,小分子化合物可以通过生物分子传感器进行胞内实时检测和分析[25]。
作者首先构建了丙二酰辅酶A 的生物传感器,实现了丙二酰辅酶A 的快速、可视化检测。 随后在大肠杆菌中构建了CRISPRi/ddCpf1 系统,并尝试将ddCpf1 和Gp2 蛋白融合。最终通过CRISPRi/ddCpf1多靶点下调脂肪酸等代谢途径,实现了丙二酰辅酶A 的积累, 为解决其下游目标产物生物合成的瓶颈问题提供参考。
1 材料与方法
1.1 材料与试剂
1.1.1 菌株与质粒大肠杆菌DH5α 用于载体构建,大肠杆菌BL21(DE3)用于生物传感器实验,由作者所在实验室保藏;Stable Competent E. coli 购于美国New England Biolabs 公司, 用于CRISPRi/ddCpf1 系统功能验证和优化;pS7a-acc、pBVSC 为作者所在实验室保存质粒,pAJK-RFP 质粒为作者构建。
1.1.2 实验试剂胰蛋白胨、 酵母提取物: 英国Oxoid 公司产品;GeneRular 1kb DNA Ladder、 琼脂糖凝胶回收试剂盒、Phusion Polymerase: 美国Thermo Fisher Scientific 公司产品;10× TAE buffer:大连美仑生物技术有限公司产品;卡那霉素、甘油、氯霉素:生工生物工程上海(股份)有限公司产品;L-阿拉伯糖: 上海麦克林生化科技有限公司产品;氯化钠、琼脂粉、葡萄糖:国药集团化学试剂有限公司产品;2×ES Taq Mastermix(Dye):北京康为世纪生物科技有限公司产品;GeneRed 核酸染料、质粒小提试剂盒:天根生化科技(北京)有限公司产品;10×DNA Loading buffer:南京诺唯赞生物科技有限公司产品;BSA(牛血清白蛋白):德国Sigma 公司产品;T4 连接酶、Dpn I 、Bsa I 限制性核酸内切酶: 美国New England Biolabs 公司产品。
1.2 方法
1.2.1 试剂配制10 g/L 氯霉素(Cm)溶液:称取0.1 g Cm 粉末溶于10 mL 的无水乙醇中, 于-20 ℃避光保存备用。
100 g/L 氨苄青霉素(Amp)溶液:称取0.2 g 粉末溶于2 mL 的ddH2O 中,过滤除菌,于-20 ℃保存备用。
50 g/L 卡那霉素(Kan)溶液:称取0.1 g Kan 粉末溶于2 mL 的ddH2O 中,过滤除菌,于-20 ℃保存备用。
0.5 mol/L 异丙基-β-D-硫代半乳糖苷(IPTG)溶液: 称取1.19 g IPTG 加入适量的灭菌ddH2O,定容至10 mL,过滤除菌,于-20 ℃避光保存。
LB 培养基:5 g 酵母提取物、10 g NaCl、10 g 胰蛋白胨, 加入去离子水搅拌混匀, 用量杯定容至1 L,高温高压灭菌20 min。 添加12 g/L 琼脂粉的LB培养基即为固体培养基。
1 mol/L 阿拉伯糖(Ara)溶液:称取0.75 g Ara粉末,加终体积为5 mL 的ddH2O 溶解,过滤除菌,4℃低温保存。
1.2.2 载体、引物和菌株构建载体采用Oligo anneal 和Golden-gate 克隆方法进行构建,构建模板来自枯草芽孢杆菌(Bacillus subtilis)、BglBrick 系列质粒[26]和Addgene 网站购买质粒。 引物合成于北京擎科生物科技有限公司上海分公司。 采用电转化法将构建好的载体转入大肠杆菌,挑取单克隆,获得目的菌株。
1.2.3 荧光值的测定将菌种置于3 mL 的LB 液体培养基(20 mg/L Kan 溶液、25 mg/L Cm 溶液、100 mg/L Amp 溶液, 抗生素添加种类根据具体菌株调整) 中220 r/min 条件下37 ℃振荡培养过夜。次日,将体积分数2%的种子液转接于3 mL 的LB 液体培养基(含相应的抗生素),振荡培养,在OD600为0.4~0.6 加入诱导剂,一定时间内吸取150 μL 或200 μL培养液于酶标仪中进行荧光值和OD600的测量。 对于ddCpf1 功能验证,OD600为0.6 时加入10 mmol/L Ara 溶液,2 h 后加入0.3 mmol/L IPTG, 振荡培养4.5 h,吸取200 μL 的培养液于酶标仪中进行测定。酶标仪设定程序: 发射波长612 nm, 激发波长560 nm。 相对荧光值的计算公式如下:

式中:F/OD600为相对荧光值;Fm为实验组荧光值;ODm为实验组OD600;Fb为LB 液体培养基的荧光值;ODb为LB 液体培养基的OD600。
2 结果与分析
2.1 丙二酰辅酶A 生物传感器的设计与构建
为了对大肠杆菌内的丙二酰辅酶A 进行方便检测,设计构建丙二酰辅酶A 生物传感器,通过红色荧光测量评估丙二酰辅酶A 的胞内浓度。 FapR蛋白来自枯草芽孢杆菌,可以特异性结合丙二酰辅酶A[25]。 如图1(a)所示,以J23119 启动子为改造基础,在-35、-10 区域前后插入FapR 蛋白结合位点,设计响应丙二酰辅酶A 浓度的启动子。 当胞内丙二酰辅酶A 浓度较低时,FapR 蛋白结合启动子,阻碍红色荧光蛋白 (red fluorescent protein,RFP) 表达;当胞内丙二酰辅酶A 浓度较高时,FapR 蛋白结合游离的丙二酰辅酶A,RFP 正常表达,可检测到显著红色荧光。 在此,设计了7 种不同FapR 蛋白结合位点的插入组合,见表1。 随后,构建丙二酰辅酶A 生物传感器(pAJK-RFP 质粒),用阿拉伯糖启动子控制FapR 蛋白组成型表达, 用丙二酰辅酶A 响应启动子控制RFP 表达,见图1(b)。

图1 丙二酰辅酶A 生物传感器原理图及质粒Fig. 1 Diagram and the plasmid of malonyl-CoA biosensor

表1 丙二酰辅酶A 响应启动子的序列Table 1 Sequences of malonyl-CoA responsive promoters
2.2 丙二酰辅酶A 生物传感器的评估
为了验证丙二酰辅酶A 生物传感器的功能,采用pS7a-acc 质粒过表达acc 基因促进丙二酰辅酶A 的积累,见图2。 将pAJK-RFP、pS7a-acc 质粒(见图1(b)和图2(b)) 转化E. coli BL21(DE3),分别采用0.1 μmol/L、1 μmol/L、10 μmol/L、100 μmol/L、1 mmol/L、10 mmol/L 的IPTG 诱导ACC 过表达,逐步使胞内的丙二酰辅酶A 浓度增加,从而开启生物传感器的荧光信号。 通过检测红色荧光值和OD600,计算生物传感器在不同IPTG 诱导浓度下的相对荧光值, 获得丙二酰辅酶A 生物传感器的响应曲线(见图3)。 J12 生物传感器渗漏控制较好, 在没有IPTG 的时候无显著荧光, 在100 μmol/L 以上的IPTG 存在时,可检测到显著的红色荧光,相对荧光值提高了3.26 倍;J233 生物传感器的荧光响应较为明显,在10 mmol/L IPTG 存在时,相对荧光值提高了5.83 倍。 后续实验选择J233 生物传感器进行。

图2 丙二酰辅酶A 生物传感器的功能验证Fig. 2 Functional verification of malonyl-CoA biosensor
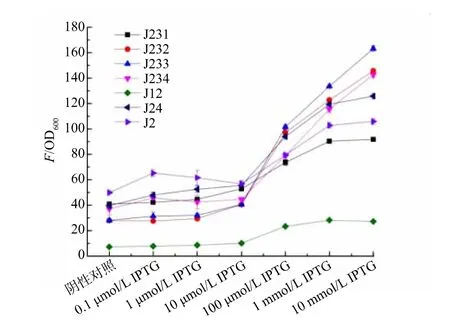
图3 不同丙二酰辅酶A 生物传感器的响应曲线Fig. 3 Response curves of different malonyl-CoA biosensors
2.3 CRISPRi/ddCpf1 系统的构建
为了构建催化失活的Cpf1, 构建了来自Francisella novicida 的Cpf1 效应蛋白的3 个突变体, 分别是D917A 单突变、E1006A 单突变和D917A-E1006A 双突变。 为了验证CRISPRi/ddCpf1系统的功能及特点, 针对PTrc启动子诱导型表达的RFP 基因设计了4 个crRNA (见图4):p-T、p-NT、R-T、R-NT,并分别靶向PTrc启动子的模板链、非模板链和靶向RFP 基因的模板链、非模板链。 以荧光数值来判断CRISPRi/ddCpf1 系统的效果。

图4 基于CRISPRi/ddCpf1 的基因抑制设计Fig. 4 Design of gene inhibition based on CRISPRi/ddCpf1
结果显示,当crRNA 靶向启动子区域时,p-T1、p-NT1 的相对荧光值在ddCpf1-M1 实验组中分别下降了89.7%和88.0%, 在ddCpf1-M2 实验组中分别下降了79.7%和87.8%, 在ddCpf1-M1-M2 实验组中分别下降了91.8%和87.0%(见图5)。 可见,突变效应蛋白ddCpf1-M1、ddCpf1-M2 和ddCpf1-M1-M2 对于p-T1、p-NT1 有较为明显的基因下调效果,且无论靶向模板链或非模板链均能得到较好的抑制效果,这与以前报道的研究结果一致[20]。当crRNA靶向RFP 基因,R-T1、R-NT1 的相对荧光值在ddCpf1-M1 实验组中分别下降了47.6%和2.2%,R-T1 的相对荧光值在ddCpf1-M1-M2 实验组中下降了57.4%(见图5)。 可见, 靶向RFP 基因时ddCpf1 的下调效率均小于靶向启动子区域时的下调效率。 对于CRISPRi/ddCpf1 系统,crRNA 靶向基因表达的启动子区域可以获得较好的基因下调效果。

图5 CRISPRi/ddCpf1 的基因抑制Fig. 5 Gene inhibition by CRISPRi/ddCpf1
2.4 CRISPRi/ddCpf1-Gp2 系统的构建
在应用中, 将dCas 蛋白融合其他功能蛋白是一个常见的改进策略。 有研究者利用dCas9 融合蛋白构建的CRISPRoff 蛋白可以持久且特异性地沉默基因表达[27];也有研究者融合dCas9 和转录抑制子(如Kox1 的KRAB 结构域), 在人类和酵母细胞中实现稳定有效的转录抑制, 揭示了dCas 融合蛋白在CRISPR 功能优化中的潜力[28]。 作者尝试将功能蛋白和ddCpf1 蛋白融合, 优化CRISPRi/ddCpf1 系统的调控功能。
研究发现,噬菌体基因2 编码的Gp2 蛋白可以降低RNA 聚合酶对启动子区域的DNA 亲和力,实现转录抑制[29]。受此启发,为了进一步提高转录抑制效率,将ddCpf1-M1 和Gp2 抑制蛋白进行融合表达(见图6(a))。 选择来自Enterobacteria phage BA14、Enterobacteria phage T3、Enterobacteria phage K1F和Klebsiella phage K11[30]的Gp2 蛋白,通过连接子(Ala-Ala,AA), 分别在ddCpf1-M1 的N 端和C 端进行融合表达(CRISPRi/ddCpf1-Gp2),见图6(b)。

图6 CRISPRi/ddCpf1 关于Gp2 蛋白的基因转录抑制Fig.6 Transcriptional inhibition of CRISPRi/ddCpf1-Gp2
结果表明, 对于Enterobacteria phage BA14 的Gp2 蛋白,连接顺序为Gp2-AA-ddcpf1-M1 的实验组的相对荧光值下降了36.1%、OD600下降了47.4%,ddcpf1-M1-AA-Gp2 实验组的相对荧光值下降了 30.3% 、OD600下降了 57.3% ; 对于Enterobacteria phage T3 的Gp2 蛋白,ddcpf1-M1-AA-Gp2 实验组的相对荧光值下降了19.5%、OD600下降了63.5%; 对于Enterobacteria phage K1F 的Gp2 蛋白,Gp2-AA-ddcpf1-M1 实验组的相对荧光值下降了37.4%、OD600下降了44.9%,ddcpf1-M1-AA-Gp2 实验组的相对荧光值下降了24.2%、OD600下降了34.2%;对于Klebsiella phage K11 的Gp2 蛋白,Gp2-AA-ddcpf1-M1 实验组的相对荧光值下降了21.0%、OD600下降了51.9%,ddcpf1-M1-AA-Gp2实验组的相对荧光值下降了30.8%、OD600下降了65.1%(见图7)。 可见,4 种Gp2 蛋白均有一定抑制基因转录的功能, 但表达Gp2 蛋白的实验组中,OD600均显著降低。 这可能是因为Gp2 蛋白的表达产生了毒性,在一定程度上干扰了大肠杆菌的生长和代谢,导致CRISPRi 的低效。
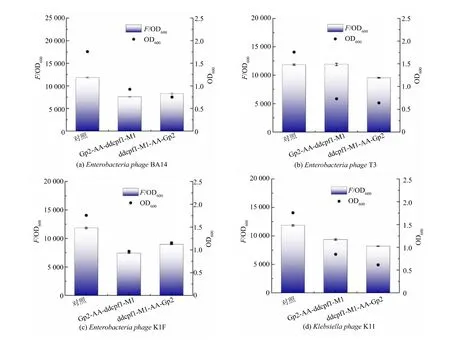
图7 4 种Gp2 蛋白的基因抑制结果Fig. 7 Gene suppression of four Gp2 proteins
2.5 通过CRISPRi/ddCpf1 技术促进大肠杆菌中丙二酰辅酶A 的积累
CRISPR 干扰可以下调基因的转录, 从而调控基因表达。为了促进大肠杆菌中丙二酰辅酶A 的合成,利用ddCpf1 双突变体(D917A-E1006A) 构建了CRISPRi/ddCpf1 双靶点系统,设计了CRISPR array( 见表2) 同时靶向两个基因:adhE ( 编码acetaldehyde dehydrogenase, 乙醛脱氢酶) 基因、fabF (编码3-oxoacyl-acyl carrier protein synthase II,3-氧代酰基-酰基载体蛋白合酶II) 基因和fabB(编码3-oxoacyl-acyl carrier protein synthase I,3-氧代酰基-酰基载体蛋白合酶I )基因、sucC(编码succinyl-CoA synthetase,琥珀酰辅酶A 合成酶) 基因[1],并进行CRISPR 干扰。通过检测RFP 相对荧光值,对胞内丙二酰辅酶A 进行测量,见图8(a)。 其中,CRISPRi/ddCpf1 系统由载体pBVSC 表达,丙二酰辅酶A 生物传感器由载体pAJK-J233 表达。
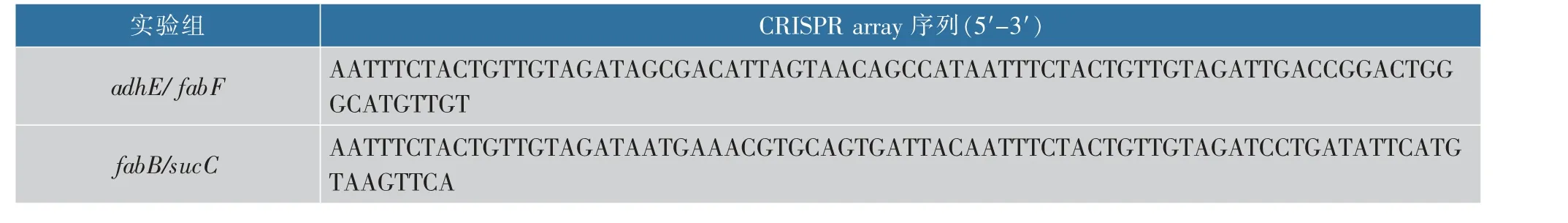
表2 CRISPRi/ddCpf1 基因转录抑制系统的CRISPR array 序列Table 2 Sequences of CRISPR array in CRISPRi/ddCpf1 gene transcriptional inhibition system
如图8(b)所示,相对于对照组(丙二酰辅酶A生物传感器菌株,不加诱导剂),CRISPRi/ddCpf1 系统靶向adhE/fabF 基因时相对荧光值增加了46.2%, 靶向fabB/sucC 基因时相对荧光值增加了149.5%。 可见,CRISPRi/ddCpf1 多重靶向策略显著促进了丙二酰辅酶A 的积累。

图8 CRISPRi/ddCpf1 促进丙二酰辅酶A 的积累Fig. 8 CRISPRi/ddCpf1 promotes the accumulation of malonyl-CoA
3 结 语
丙二酰辅酶A 是重要的前体物质,其合成被认为是大规模生产目标次级代谢产物的潜在瓶颈。 研究表明,其常规基因敲除会影响细胞生长。 因此,利用CRISPR 干扰技术实现基因转录下调, 从而促进丙二酰辅酶A 的积累。为了比较丙二酰辅酶A 的浓度,作者以J23119 启动子为改造基础,构建了7 个不同的丙二酰辅酶A 生物传感器,通过红色荧光值的测定实现了丙二酰辅酶A 的快速、 可视化测量。然后,构建了CRISPRi/ddCpf1 基因干扰系统,成功实现了目的基因的基因沉默,并比较了不同ddCpf1突变体、 不同crRNA 靶向位置对基因沉默的影响。随后, 尝试融合Gp2 和ddCpf1 效应蛋白对该CRISPRi/ddCpf1 系统进行优化,但基因转录下调效率没有显著提升。 最后,利用CRISPRi/ddCpf1 技术同时靶向两个基因(adhE、fabF 和fabB、sucC) 进行CRISPR 干扰,显著促进了丙二酰辅酶A 的积累,为其下游目的产物的高效合成提供了技术参考。 在后续研究中, 需要尝试融合其他蛋白、 设计CRISPR array 进行多重基因抑制等,实现对基因转录下调效率的控制。
猜你喜欢
——一道江苏高考题的奥秘解读和拓展
中学生物学(2022年7期)2022-09-07
中老年保健(2022年4期)2022-08-22
成都医学院学报(2022年4期)2022-08-19
中老年保健(2022年1期)2022-08-17
保健医苑(2022年5期)2022-06-10
江西农业学报(2021年4期)2021-04-20
三农资讯半月报(2020年11期)2020-06-21
华声文萃(2019年4期)2019-09-10
文萃报·周二版(2019年10期)2019-09-10
中华建设(2019年7期)2019-08-27
