
不同米蛋白组分与铅的结合规律
2022-01-07 08:52范大明董田田李克强倪双双王韧
食品与生物技术学报 2021年12期
冯 伟, 范大明, 董田田, 李克强,倪双双, 王 涛, 张 昊, 王韧*
(1. 粮食发酵工艺与技术国家工程实验室, 江南大学, 江苏无锡 214122; 2. 江南大学食品学院, 江苏无锡 214122)
大米是我国重要的主粮,近年来大米重金属污染事件频发,对国民健康造成了潜在危害[1]。 铅(Pb2+)是大米重金属污染中最典型的元素之一,Pb2+由工业“三废”排放、矿藏开采等途径进入土壤和水源,随着水稻生长富集,再通过饮食进入人体,人体中血铅超标易引起贫血, 损害人体神经、 血液、骨骼、消化、生殖等系统[2]。
Pb2+主要与大米中的蛋白质组分相结合[3]。 现有研究工作主要侧重于Pb2+在稻米中的分布规律、检测技术、脱除方法、健康风险评估等方面[4-6],例如张园园等采用气浴振荡辅助柠檬酸法脱除稻谷中的Pb2+,在优化条件下Pb2+脱除率达34.27%[6],但从理论层面探索Pb2+与蛋白质的结合特征、 结合机理的报道却鲜有见闻。 根据在不同溶剂中溶解度的差异,米蛋白各组分分为清蛋白、球蛋白、醇溶蛋白和谷蛋白,其中清蛋白水溶性好,且主要分布于米糠中[7],研究其与Pb2+的结合代表性不强。
作者以3 种水不溶性米蛋白组分 (球蛋白、醇溶蛋白和谷蛋白)为研究对象,通过建立动力学模型和等温吸附模型探究各组分与Pb2+的结合机制,并通过X 射线光电子能谱(XPS)和扫描电镜(SEM)对与Pb2+结合后的醇溶蛋白进行表征, 进一步探讨蛋白质与Pb2+的结合位点及蛋白质微观结构变化,以期为大米除铅技术的开发提供理论依据。
1 材料与方法
1.1 材料与试剂
大米:稻米品种为南粳9108,泰州市和平米业有限公司提供,采用衢州市库米赛诺粮食机械制造有限公司的高性能节能砂带碾米机 (KMSNMNMD65-B)经4 道碾磨加工制备而成;硝酸铅、氢氧化钠、盐酸、硫酸、氯化钠、氯化铁、氯化钙、乙醇、溴化钾、乙二胺四乙酸二钠、柠檬酸三钠、醋酸钠、焦磷酸钠(分析纯)、硝酸(优级纯):国药集团化学试剂有限公司产品。
1.2 仪器与设备
Avanti J-26 XP 高速离心机: 美国贝克曼公司产品;透析袋(截留相对分子质量50 000):上海哈灵生物科技公司产品;Beta 2-8 L Dplus 型冻干机:德国Marin Christ 公司产品;X-射线光电子能谱分析仪250 XI: 日本岛津公司产品;Quanta 200 型扫描电子显微镜: 美国FEI 公司产品;WX-6000 型微波消解仪: 上海屹尧仪器科技发展有限公司产品;AA-220/220Z 原子吸收分光光度计:美国瓦里安公司产品;Millipore-Q 型超纯水系统: 美国Millipore公司产品;RV10 型旋转蒸发仪:德国IKA 公司产品。
1.3 实验方法
1.3.1 米蛋白组分的制备3 种水不溶性米蛋白的制备参照传统Osborne 法并加以改良[8],如下:将大米粉碎后过80 目筛, 按固液比1 g∶8 mL 混合大米粉和去离子水,用1.0 mol/L NaOH 调节pH 至12.0,搅拌提取4 h 后5 000 g 离心10 min 收集上清液。用1.0 mol/L HCl 调节上清液pH 至4.5,5 000 g 离心10 min 收集沉淀,即为米蛋白组分混合物。 将此蛋白质混合物依次用去离子水、4 g/dL NaCl、 体积分数70%的乙醇、0.1 mol/L NaOH 连续提取, 各搅拌1 h 后5 000 g 离心10 min,弃去水提上清液,收集其余步骤上清液。 用0.1 mol/L HCl 调节盐提、碱提上清液pH 分别至4.3 和4.8,5 000 g 离心10 min收集相应沉淀。 醇提上清液采用旋转蒸发仪40 ℃蒸发浓缩至原体积的10%, 加入10 倍去离子水混匀,5 000 g 离心10 min 收集沉淀。将上述3 种沉淀分别用去离子水调成pH 6.0 的溶液,置于透析袋中透析24 h,每4 h 更换去离子水。透析结束进行冷冻干燥,得到球蛋白、谷蛋白和醇溶蛋白,样品放置于-22 ℃冰箱备用。
1.3.2 米蛋白与Pb2+的结合反应准确称取0.06 g米蛋白,加入10 mL 超纯水,水浴振荡分散1 h,按照设定的Pb2+质量浓度,加入一定体积硝酸铅溶液,用超纯水补至20 mL, 在30 ℃水浴中以150 r/min振荡反应一定时间,反应结束后8 000 g 离心3 min取上清液,采用火焰原子吸收分光光度计测定吸附前后溶液中Pb2+质量浓度。 米蛋白的Pb2+结合量(q)可参照下方公式计算。
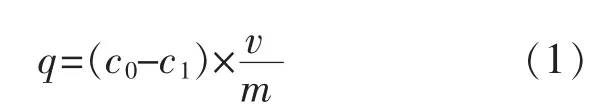
式中:q 为米蛋白的Pb2+结合量,mg/g;c0为溶液中Pb2+的初始质量浓度,mg/L;c1为反应后溶液中Pb2+质量浓度,mg/L;m 为米蛋白质量,g;v 为溶液体积,L。
1.3.3 理化指标测定
1) Pb2+质量浓度的测定 参照GB 5009.12—2017《食品安全国家标准食品中铅的测定》中第三法火焰原子吸收光谱法。
2) 蛋白质的测定 参照GB 5009.5—2016《食品安全国家标准食品中蛋白质的测定》中第一法凯式定氮法,氮转化系数为5.95。
1.3.4 动力学模型建立参照1.3.2 所述方法,设定Pb2+初始质量浓度为100 mg/L,pH 6.0,反应温度30 ℃, 在反应时间分别为2、5、10、30、60、120 min时测定溶液中Pb2+质量浓度, 得到q 随时间的变化曲线,采用准一级动力学模型(公式2)、准二级动力学模型(公式3)以及Elovich 动力学模型(公式4)对数据进行拟合[9]。
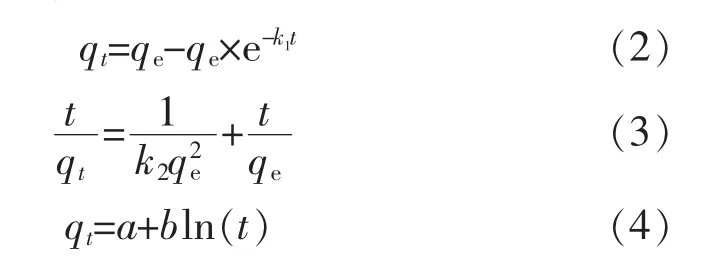
式中:qe为平衡吸附量,mg/g;t 为反应时间,min;k1为准一级动力学反应速率常数,min-1;k2为准二级动力学反应速率常数,g/(mg·min);a,b 为反应常数。
1.3.5 等温吸附模型的建立参照1.3.2 所述方法, 设定Pb2+初始质量浓度分别为10、20、40、60、80、100、120、140 mg/L,pH 6.0, 反应时间120 min,并分别在15、30、45 ℃下进行反应, 绘制等温吸附曲线,采用Langmuir 模型(公式5)和Freundlich 模型(公式6)进行拟合[10],其线性化方程如下:
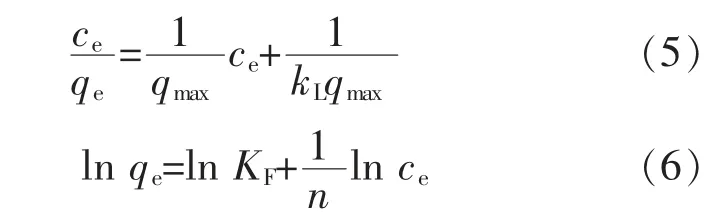
式中:qe为平衡吸附量,mg/g;qmax为饱和吸附量,mg/g;ce为平衡质量浓度,mg/L;KL为与吸附强度有关的L模型吸附平衡常数;KF为与吸附容量有关的F 模型吸附平衡常数;n 为与吸附强度有关的F 模型常数。
参照文献[11-12], 热力学参数吉布斯自由能ΔG°(kJ/mol)、焓变ΔH°(kJ/mol)和熵变ΔS°(J/mol)的计算采用以下公式:
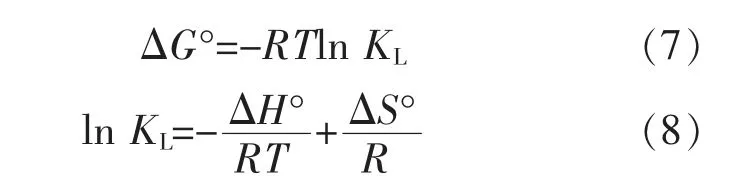
式中:KL为L 模型吸附平衡常数;R 为理想气体常数,8.314 J/(mol·K);T 为开尔文温度,K。
1.3.6 X 射线光电子能谱分析(XPS)醇溶蛋白在Pb2+初始质量浓度100 mg/L, 温度30 ℃,pH 6.0 条件下反应2 h,反应结束后离心取沉淀,水洗2 次,然后进行冷冻干燥。 将干燥的醇溶蛋白和醇溶蛋白-Pb2+结合物用于XPS 分析。 使用Al-KαX 射线源, 能谱扫描范围为1~1 300 eV, 用结合能为285.0 eV 的C1s 校准光谱进行校正。
1.3.7 表观形态分析将上述干燥后的醇溶蛋白和醇溶蛋白-Pb2+结合物固定于导电胶表面,在真空环境中喷涂铂金(厚度约30 nm),使用Quanta-200扫描电子显微镜(SEM)观察拍摄。
1.4 数据处理
利用SPSS 17.0 和Excel 软件进行数据处理及统计分析。利用Origin 9.0 软件进行图形绘制。数据以平均值±标准差的方式表示。 每次实验重复3 次。
2 结果与讨论
2.1 不同米蛋白样品的蛋白质质量分数
采用改良Osborne 法提取得到的3 种米蛋白质量分数见表1,其中球蛋白与谷蛋白达到94%以上,醇溶蛋白达到88%,质量分数较高。 传统方法中蛋白质质量分数较低,主要杂质是淀粉及多糖,而作者采取的方法在第一步总蛋白的提取中已除去了大量的杂质,且经过水洗、透析等步骤,纯度得到很大提高。
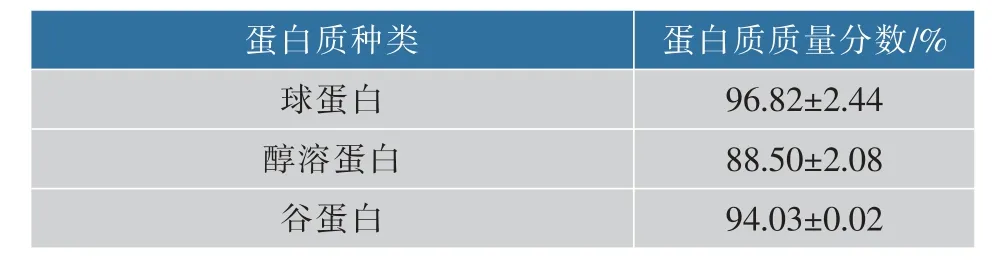
表1 3 种米蛋白的质量分数分析Table 1 Mass ratio analysis of different rice protein
2.2 不同米蛋白与Pb2+结合的动力学
如图1 所示,球蛋白、醇溶蛋白、谷蛋白3 种蛋白质与Pb2+的结合均在较短时间内达到了平衡,在前10 min 内反应迅速,之后反应速度变慢并逐渐达到饱和,30 min 后q 基本不再增加。 这可能是因为蛋白质表面可结合Pb2+的位点数有限, 反应开始时大量Pb2+急速涌向结合位点,导致q 快速升高;随着Pb2+逐渐结合到蛋白质上, 蛋白质结构的致密性增加,结合位点被占据殆尽,q 增速越来越缓慢[13],并逐渐达到平衡状态。 醇溶蛋白、球蛋白、谷蛋白的平衡结合量依次为20.54、12.00、3.32 mg/g, 可见醇溶蛋白对Pb2+的吸附能力最强。

图1 不同米蛋白与Pb2+的结合量(q)随时间的变化曲线Fig. 1 Adsorption quantity (q) of different rice protein to Pb2+at the different time
如表2 所示,采用准一级动力学模型、准二级动力学模型以及Elovich 动力学模型来描述3 种米蛋白与Pb2+的结合过程。 准一级动力学模型假设吸附速率与有效吸附位点数成正比, 即为物理吸附;准二级动力学模型假设吸附速率与有效吸附位点数的平方成正比,吸附质与吸附剂表面的官能团之间发生了电子共享或者交换,即为化学吸附[14-15];Elovich 动力学模型综合了准一级和准二级动力学模型的边界条件范围,即同时存在物理吸附与化学吸附[16]。表2 中Elovich 动力学模型和准一级动力学模型拟合得到的线性相关系数R2较低,准二级动力学模型的拟合参数R2均达到0.99 以上, 且拟合得到的该反应条件下的理论平衡吸附量(qe)与前文中的实测值十分接近,说明准二级动力学模型可以更好地拟合3 种米蛋白与Pb2+的结合过程, 此结合反应以化学吸附为主。
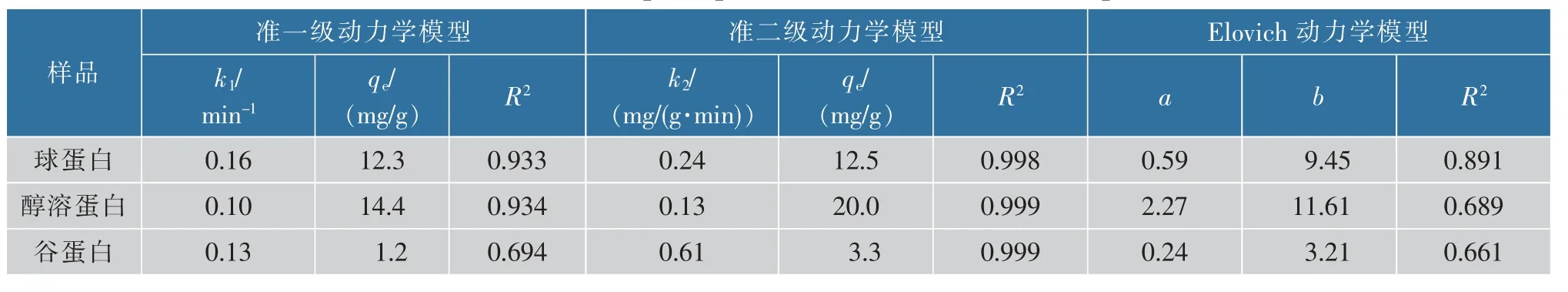
表2 不同米蛋白与Pb2+结合的动力学参数Table 2 Kinetic adsorption parameters of Pb2+to different proteins
2.3 不同米蛋白与Pb2+结合的等温吸附模型及热力学分析
图2 显示了在不同初始质量浓度、 不同温度下,Pb2+平衡质量浓度与各蛋白质组分qe的关系。当Pb2+质量浓度较低时,qe增长较快, 尤其是醇溶蛋白;而当Pb2+质量浓度较高时,qe的增长逐渐变缓并趋稳。 这可能是因为蛋白质表面可与Pb2+结合的位点数量一定, 低初始质量浓度时Pb2+可结合的位点充足,结合迅速[17];随着Pb2+质量浓度的增加,蛋白质表面未被占据的结合位点逐渐减少, 同时高Pb2+质量浓度导致蛋白质发生聚集,比表面积减小[18],结合位点总量进一步减少,促进反应趋向平衡。除Pb2+初始质量浓度的影响之外, 温度对蛋白质与Pb2+的结合也有重要影响,3 种米蛋白均呈现出温度升高利于吸附的现象, 这与Liu 关于大豆微球蛋白对Pb2+吸附的研究结论一致[19]。 在一定范围内温度上升,一方面可促进蛋白质溶胀,使肽链伸展,进而比表面积增大,基团暴露,有利于吸附的进行;另一方面也可能是由于各蛋白质组分与Pb2+的结合过程为吸热反应,适当升温提高了反应的平衡常数。
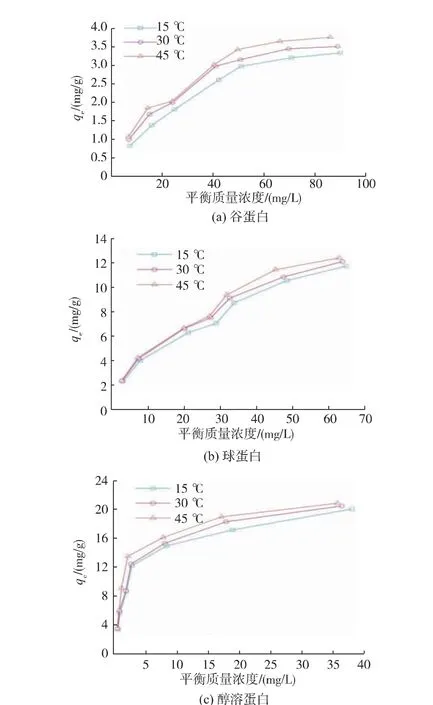
图2 不同米蛋白与Pb2+结合的等温吸附线Fig. 2 Isothermal adsorption line of Pb2+binding to different proteins
采用Langmuir 模型和Freundlich 模型对3 种米蛋白在不同温度下的吸附数据进行拟合,见表3。相比于Freundlich 模型,Langmuir 模型的R2较高,可以更好地拟合蛋白质与Pb2+的结合过程。Langmuir 模型主要适用于发生在吸附基质表面的单分子层吸附过程,该模型假定吸附基质表面均匀分布着活性位点且不发生相互作用[20]。 这说明3 种米蛋白与Pb2+的结合均是以均匀的单分子层吸附为主,这也解释了图1 中q 随时间变化快速达到平衡的原因。 此外,表3 中3 种米蛋白的最大吸附量qmax均随着温度的升高而增大。 吸附反应过程一般同时存在物理吸附和化学吸附,物理吸附是以范德华力为作用力的单分子层或多分子层吸附,此类吸附不稳定、易解吸、不受温度影响;化学吸附则是以化学键为作用力的单分子层吸附,温度升高可加快吸附速率,一定范围内可提高吸附量[21]。由此推断3 种米蛋白组分与Pb2+的结合主要为化学吸附, 这与前文中动力学分析的结论一致。
由表3 中的热力学数据可知,3 种米蛋白的ΔG°均为负值, 表明3 种米蛋白与Pb2+的结合是自发反应;ΔS°、ΔH°均为正值,说明此结合过程是熵驱动的吸热反应,进一步证实了前文中关于随温度升高qmax增大原因的推测。 从数值来看,3 种米蛋白与Pb2+结合的ΔH°较小,且为正值,ΔS°为正值,这说明可能是疏水作用在吸附过程中起到了重要作用[22]。醇溶蛋白的ΔS°、ΔH°数值均高于其他两种蛋白质,说明醇溶蛋白对Pb2+有较强的吸附性能[23]。
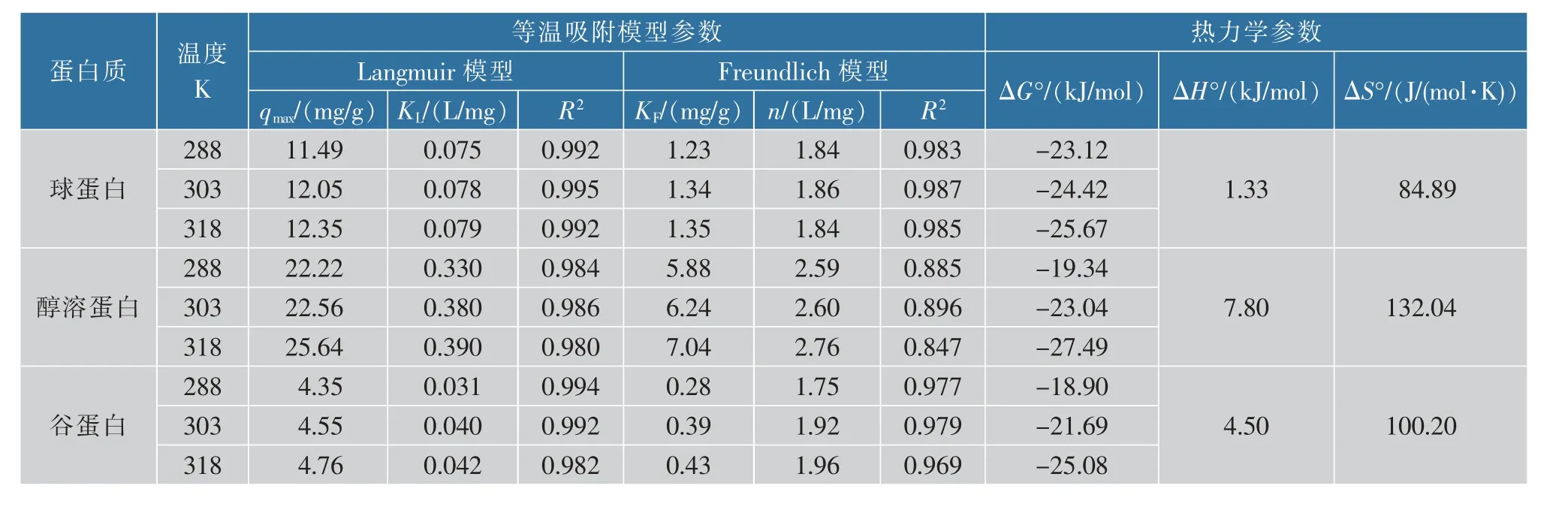
表3 不同米蛋白与Pb2+结合的等温吸附模型及热力学参数Table 3 Isothermal adsorption models fitting parameters and adsorption thermodynamic parameters of Pb2+ to different proteins
2.4 醇溶蛋白与Pb2+结合的基团变化
采用X 射线光电子能谱技术(XPS)对醇溶蛋白与Pb2+结合后的基团变化进行表征。 如图3 (a)所示,在140 eV 附近观察到Pb4f 结合能的特征峰[24],说明Pb2+与醇溶蛋白之间发生了化学结合。 S 原子的峰发生了明显的重叠现象,故采用Peakfit 软件通过高斯拟合进行分峰处理,获取单峰信息。 图3(c)显示, 醇溶蛋白O1s 在532.3 eV 附近有显著的峰值,且峰位移较小、峰强度减弱,这个位置对应于醚基团(主要是羧基)[11,25]。醇溶蛋白N1s 结合Pb2+后在399.9 eV 处出峰,结合能未发生转移但强度显著降低(P<0.05),说明含N 基团与Pb2+发生了结合[26]。 醇溶蛋白S2p 分别在159.8、163.8 eV 和167.9 eV 附近出峰,代表硫醚基(—S—)、巯基(—SH)和磺酸基(—SO3)[27],这些基团结合能的移动或强度变化均说明了含硫基团与Pb2+发生结合作用。Takashi 等在研究中发现,大米醇溶蛋白中含硫的氨基酸含量较高,这正是醇溶蛋白对Pb2+吸附能力强的原因[28]。
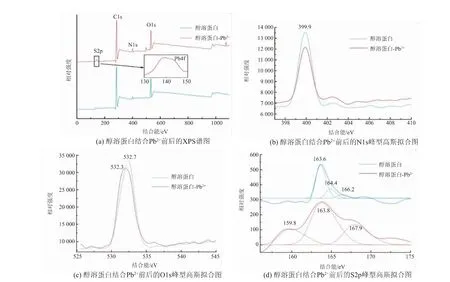
图3 醇溶蛋白结合Pb2+前后的XPS 及峰型高斯拟合图Fig. 3 XPS spectra of prolamin before and after binding with Pb2+
2.5 醇溶蛋白与Pb2+结合的表观形态变化
醇溶蛋白结合Pb2+前后的扫描电子显微镜形貌观察见图4, 未结合Pb2+的醇溶蛋白原本呈现出片层连接结构; 在结合Pb2+之后, 蛋白质颗粒发生聚集,并形成了更加致密的团块状结构。

图4 醇溶蛋白结合Pb2+前后的SEM 图Fig. 4 SEM image of prolamin before and after binding Pb2+
这证实了前文中关于Pb2+导致蛋白质发生聚集,引起比表面积减小、结合位点总量减少的猜测。此聚集现象主要是由于Pb2+所带正电荷与蛋白质表面带负电荷的基团形成化学键引起[18],与等温吸附模型的解释一致。
3 结 语
通过对3 种水不溶性米蛋白组分与铅的结合能力进行分析,得到如下结论:3 种蛋白质对铅的结合均是以在蛋白质表面的单分子层形式发生化学吸附为主,是熵驱动的自发吸热反应过程,结合过程符合准二级动力学模型、Langmuir 等温吸附模型。 3 种蛋白质中醇溶蛋白对铅的吸附能力最强,XPS 谱图表明醇溶蛋白与铅发生结合的基团主要是含氮、含硫基团,蛋白质结构由未结合时的片层状转变为结合后的聚集团块状, 结构更加致密,引起结合位点总量减少,可加快反应平衡的速度。
猜你喜欢
煤气与热力(2021年12期)2022-01-19
上海金属(2021年6期)2021-12-02
大连民族大学学报(2021年5期)2021-11-15
昆明医科大学学报(2021年3期)2021-07-22
环境卫生工程(2021年3期)2021-07-21
烟草科技(2021年6期)2021-06-24
当代化工(2019年3期)2019-12-12
新课程·下旬(2019年7期)2019-09-17
电脑知识与技术(2018年19期)2018-11-01
发明与创新·中学生(2017年11期)2017-12-07
