
红皮病性大疱性类天疱疮1例
2022-01-04 10:41张亚利李小红杨秋萍张曼曼张江安于建斌
皮肤性病诊疗学杂志 2021年6期
张亚利, 李小红, 杨秋萍, 张曼曼, 张江安, 于建斌
郑州大学第一附属医院皮肤科,河南 郑州 450052
1 临床资料
患者女,78岁。因“躯干、四肢红斑、鳞屑伴瘙痒5年,加重3年余”就诊于我院皮肤科。5年前患者无明显诱因躯干、四肢出现多发红斑、鳞屑,伴瘙痒,无丘疹、水疱、脓疱等症状,不伴发热。期间当地多家医院间断治疗,诊断不详,疗效欠佳。3年前皮损加重泛发全身,出现弥漫性红斑、浸润,大量脱屑,伴剧烈瘙痒,至当地医院按“红皮病”给予糖皮质激素治疗,具体用量不详,病情好转,数月后病情复发,表现为全身弥漫性红斑,为求进一步诊疗,至我科门诊。否认药物、食物过敏史,余既往史、个人史、家族史等无特殊。入院时皮肤科检查:全身弥漫性红斑、浸润,大量糠样鳞屑,其间可见片状正常皮肤,银屑病三征阴性,尼氏征阴性。实验室检查:尿常规:白细胞3+;血沉:41.00 mm/h;肿瘤标志物:癌胚抗原5.52 ng/mL,铁蛋白244.9 ng/mL,余均在正常范围。外周血免疫球蛋白IgE>2 500.00 IU/mL;血常规、肝肾功、血糖、电解质、心肌酶谱、凝血、风湿全套未见明显异常;胸部CT示:两肺慢性炎症,纵膈内及双侧腋窝多发增大淋巴结;组织病理(腋窝淋巴结):考虑淋巴结反应性增生。患者住院1周时,大腿及腹部出现散在紧张性水疱,疱液清亮,尼氏征阴性,水疱破裂后形成糜烂面,伴有少量渗液(图1A、1B)。住院第6天、第9天查血清抗BP180抗体均阳性,分别为103.97 U/mL和125.87 U/mL,抗BP230抗体、抗桥粒芯蛋白抗体阴性。住院1周后皮损组织病理检查可见表皮下水疱,真皮浅层大量淋巴细胞为主的浸润(图2)。直接免疫荧光(图3A~3C):基底膜带IgG和C3线状沉积,IgA阴性。诊断:红皮病性大疱性类天疱疮。住院第4天开始给予甲泼尼龙40 mg静脉滴注每天1次,治疗1周,病情好转出院,院外继续甲泼尼龙40 mg静脉滴注每天1次,出院10 d后复查,全身红斑、鳞屑减轻,水疱消退,可见色素沉着斑(图4A、4B),瘙痒消失。之后不定期复查,激素逐渐减量,2个月后减量至泼尼松片15 mg口服每天1次,病情稳定。1年后随访,患者泼尼松片15 mg口服每天1次维持治疗,躯干四肢可见轻度红斑,无水疱、糜烂和瘙痒。

图2 组织病理:可见表皮下水疱,真皮浅层以淋巴组织细胞浸润为主(HE,200×)
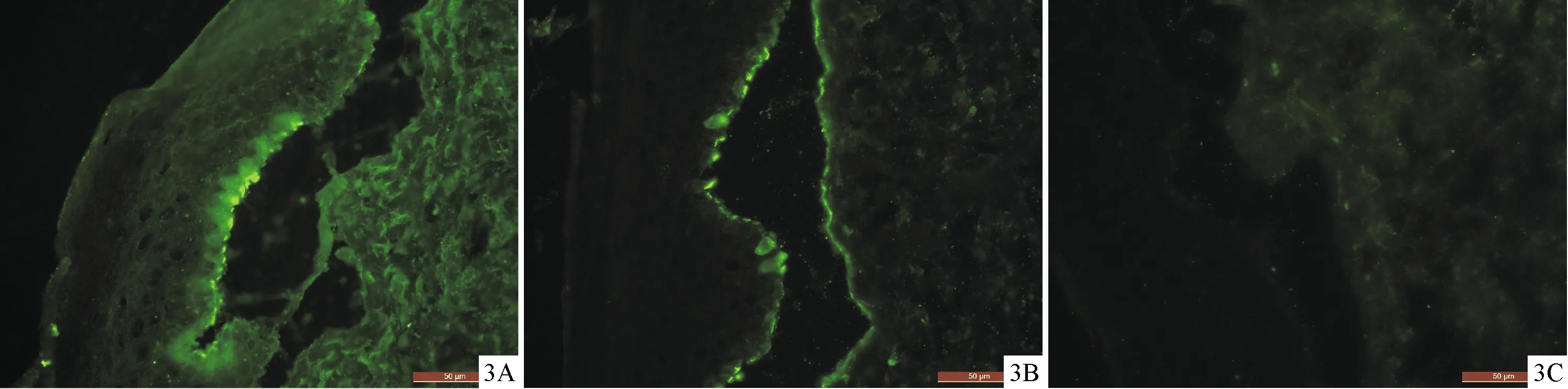
图3 直接免疫荧光基底膜带IgG(3A)、C3(3B)呈线状沉积,基底膜带IgA(3C)阴性(DIF,100×)

图4 治疗后皮损
2 讨论
大疱性类天疱疮(bullous pemphigoid, BP)是一种自身免疫性大疱性皮肤病,主要发生于老年人,目前病因和发病机制尚不清楚。大疱性类天疱疮抗原(bullous pemphigoid antigen,BPAG)位于皮肤基底膜带的半桥粒中,分别为BPAG1和BPAG2,前者的相对分子质量为23 000,是半桥粒的细胞内成分;后者的相对分子质量为18 000,是跨膜蛋白,在疾病发生中起显著作用。自身抗体与BPAG1和BPAG2相结合,使补体激活、炎症细胞募集、炎症介质和蛋白酶释放、真表皮之间的连接结构破坏,导致真皮与表皮分离,在正常皮肤或红斑基础上出现紧张性水疱[1]。

图1 患者住院1周时临床图片 1A、1B:躯干、四肢皮肤弥漫红斑、浸润和鳞屑,双下肢、腹部散在水疱和糜烂
Figure 1 Clinical pictures of the patient at 1 week of hospitalization 1A、1B:Diffused erythema, infiltration and scales scattered on the trunk and extremities, scattered blisters and erosion on the lower limbs and abdomen.
红皮病性大疱性类天疱疮(erythrodermic bullous pemphigoid, EBP)是BP的特殊亚型,临床罕见。1982年Tappeiner 等[3]首次报道了1例89岁的女性患者,表现为红皮病和散在水疱,右大腿水疱活检组织病理学诊断为BP;直接免疫荧光显示C3在皮肤基底膜带有线状沉积;间接免疫荧光显示患者血清和疱液中抗BMZ抗体滴度为1∶1 280,诊断为EBP。1993年Saitoh等[4]报道了1例76岁的日本女性患者,与本报道的病例同样有明显的IgE升高,但本例患者没有其持续高热、白细胞及嗜酸性粒细胞增多现象。此后,陆续有国外学者报道该病[5-10],包括女4例,男2例,确诊时年龄77~89岁;患者水疱皮损可出现在红皮病表现之前(4例)或之后(2例)。国内仅2010年报道过1例66岁的EBP男性患者[2]。从目前不多的国内外病例报道中得知,本病好发于60岁以上的老年人,表现为红斑、浸润、鳞屑伴不同程度瘙痒,一般无全身症状,临床极易误诊。
EBP主要诊断依据是患者临床表现为弥漫性红斑和鳞屑,伴瘙痒,病程中出现水疱,直接免疫荧光表皮基底膜带IgG和(或)C3线状沉积,血清检测到BP180和(或)BP230抗体阳性是诊断EBP的重要依据。EBP的临床鉴别诊断主要包括红皮病型银屑病、毛发红糠疹、泛发性湿疹、剥脱性皮炎型药疹、挪威疥和皮肤T细胞淋巴瘤[11-14]。红皮病型银屑病多有寻常型或脓疱型银屑病病史;毛发红糠疹具有特征性的棕红色毛囊角化性丘疹和掌跖角化过度等特点;泛发性湿疹皮损呈多形性,有渗出倾向,境界不清;剥脱性皮炎型药疹一般有明确的用药史,常由磺胺类、别嘌呤醇、卡马西平、抗癫痫药、砷剂等引起,一定潜伏期后出现红斑、浸润和鳞屑,伴瘙痒;挪威疥多发生于年老体弱、免疫力功能低下、营养不良、卫生条件差或患有严重系统性疾病、皮肤感觉障碍的患者,主要表现为角化性皮损,可出现红斑、脱屑,有特殊臭味,皮屑镜检可发现疥虫或虫卵等;皮肤T细胞淋巴瘤可表现为红皮病,组织病理特点为异型淋巴细胞嗜表皮现象和Pautrier微脓疡等。另外,其病理鉴别诊断包括天疱疮、线状IgA大疱性皮病、类天疱疮样扁平苔藓等。天疱疮中的红斑落叶型,组织病理为表皮内水疱,棘层松解,直接免疫荧光IgG和/或C3棘细胞间网状沉积。线状IgA大疱性皮病,组织病理表现为表皮下水疱,但直接免疫荧光基底膜呈线状IgA沉积。类天疱疮样扁平苔藓, 直接免疫荧光检查显示基底膜带有IgG和/或C3呈线状或颗粒状沉积,丘疹斑块处组织病理改变有扁平苔藓的特点。总之,依据典型临床表现及组织病理、免疫病理特征,可以对红皮病性大疱性类天疱疮进行诊断,但该病仅有红斑鳞屑表现时需要与多种皮肤病进行鉴别,在疾病进程中水疱表现可能出现较晚,或仅为一过性水疱,极易造成误诊。
如临床发现有类天疱疮患者表现为红皮病[15],提示对于临床以红皮病为表现,剧烈瘙痒且IgE升高者,应考虑有EBP的可能性,应进行组织病理、直接免疫荧光和血清疱病抗体检查,以及时明确诊断,避免漏诊和误诊。本例患者以散在红斑、大量脱屑为初发表现,在病程进展中逐渐发展为全身弥漫性潮红,数年后出现水疱,伴IgE显著升高。其诊断为EBP的依据主要有:①临床表现为剥脱性皮炎,全身皮肤弥漫性潮红、大量脱屑伴剧烈瘙痒,病程期间出现散在水疱;②皮损组织病理检查提示表皮下水疱,真皮上部可见淋巴细胞浸润;③直接免疫荧光表现为表皮基底膜带IgG和C3线状沉积;④血清抗BP180抗体阳性。给予甲泼尼龙治疗后病情好转。随访现仍以泼尼松片15 mg口服每天1次维持治疗,皮疹未复发,全身无红斑水疱、糜烂和瘙痒。
猜你喜欢
中国美容医学(2022年2期)2022-03-17
家庭医学·下半月(2018年8期)2018-10-17
家庭医药(2018年2期)2018-02-09
红蜻蜓(2017年6期)2017-10-30
飞碟探索(2017年8期)2017-08-11
家庭医药(2016年4期)2016-05-04
老友(2016年2期)2016-03-15
中国民族民间医药·下半月(2014年2期)2014-09-26
故事作文·低年级(2009年4期)2009-04-21
