
成人表皮痣综合征1例
2022-01-04 10:41:06刘英杜娟杨云友龙恒
皮肤性病诊疗学杂志 2021年6期
刘英, 杜娟, 杨云友, 龙恒
1.文山州皮肤病防治所,云南 文山 663099;2.复旦大学附属华山医院皮肤科,上海 200040
1 临床资料
患者男,28岁,因皮疹、智力低下、多动28年,间断抽搐27年来诊。患者出生后3个月时颈部出现灰黑色、米粒大丘疹,触之硬。此后皮损逐渐扩大并增多,遍布全身,颜色加深,无自觉症状,家长发现其智力低下、多动,走路时易跌倒。1岁因发热后首次出现间歇性癫痫大发作(以后无诱因发作),每天发作1~2次,发作后不思饮食、无力。服用“卡马西平、丙戊酸镁缓释片”1年,效果差,后改为“氨硝安定”和“奥卡西平”口服及变动生活环境(从农村到城市),抽搐次数减少(每周发作1~2次),但仍未完全控制。腋下、腹股沟有特殊腥臭味,自觉瘙痒。其父在患者5岁时因“膀胱癌”去世,母健在,其外祖父母、父母均非近亲结婚,父母、两个兄弟、血亲三代无异常。母孕期无特殊用药史及病毒感染或放射线接触史。体格检查:发育异常,早老外观、粗糙面容(似60多岁), 浅表淋巴结未触及肿大,牙发育不全、稀疏缺失,心肺腹检查未见明显异常。
皮肤科情况(图1A、1B):额部、眼周、颈部可见弥漫性密集的淡褐至褐黑色乳头瘤样角化性丘疹,触之坚硬,似高起鱼鳞病、黑棘皮病样,颈部伴有多发息肉状增生;躯干、四肢皮肤角质增厚,可见弧形带状或旋涡状排列,不规则斑片状或斑点状色素脱失斑,与增生性皮损及正常皮肤相互交错。面部、外生殖器有受累,掌跖未受累,全身毛发正常。
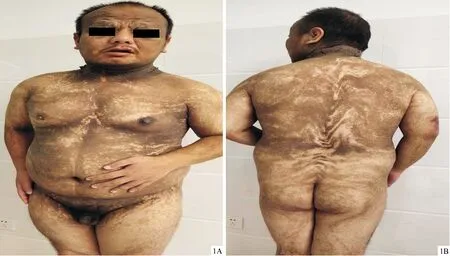
图1 患者临床图片 1A、1B:额部、眼周、颈部可见弥漫性密集的淡褐至褐黑色乳头瘤样角化性丘疹;躯干、四肢皮损角质增厚,较额部、眼周、颈部薄,可见弧形带状或旋涡状排列,不规则斑片状或斑点状色素脱失斑,与增生性皮损及正常皮肤相互交错
实验室检查:血、尿、粪便常规正常,梅毒及艾滋病检测、真菌涂片均阴性,肝肾功能正常;心脏彩超、胸部X线片、腹部彩超未见异常。头颅MR检查:双侧颞角及大脑大静脉池稍扩张,双侧海马体积稍小;双侧脑室旁白质区、双侧额叶皮层下及白质区少许缺血性损害灶。脑电图:脑电图异常伴高中波幅尖波及4~7 Hz的θ节律短阵出现。颈部皮损组织病理检查(图2A)示:表皮角化过度,棘层乳头瘤样增生,棘细胞空泡状,基底细胞层黑素颗粒增多;真皮内为疏松的结缔组织,纤维母细胞增生,其中可见较多扩张毛细血管,血管周围轻度炎细胞浸润,病理诊断:呈纤维上皮性息肉改变。胸部皮损组织病理检查(图2B)示:表皮角化过度,棘层肥厚,乳头瘤样增生,皮突不规则延长,基底细胞层黑素颗粒增多;真皮浅层血管周围少量淋巴细胞、组织细胞及噬黑素细胞浸润,偶见个别嗜酸性粒细胞,病理诊断:呈表皮痣样改变。未做遗传学基因检测。诊断:根据皮疹特点、组织病理、遗传标准可明确诊断为表皮痣综合征。

图2 皮损组织病理(HE,40×) 2A:颈部:表皮角化过度,棘层乳头瘤样增生,基底细胞层黑素颗粒增多;2B:胸部:表皮角化过度,棘层肥厚,乳头瘤样增生,皮突不规则延长,基底细胞层黑素颗粒增多
2 讨论
表皮痣综合征(epidermal nevus syndrome)是一种罕见的表皮痣伴有皮肤、眼和神经、骨骼、心血管及泌尿生殖系统发育畸形的神经皮肤综合征,又称为Schimmelpenning综合征、Feuerstein-Mimsz综合征、Solomon综合征[1-2]。1968年Solomom首次提出表皮痣综合征的概念[3]。根据临床表现、组织病理学及遗传特征的不同可分为5个亚型,包括Schimmelpenning综合征(皮脂腺痣综合征)、黑头粉刺样痣综合征、色素性表皮毛发痣综合征(Becker痣综合征)、Proteus综合征(又称变形综合征)和CHILD综合征(先天性半侧发育不良、鱼鳞病样红斑及肢体缺陷)。2004年斑痣性错构瘤病样色素沉着角化病被归到表皮痣综合征中[4]。本病也可以通过相关表皮痣的类型和有无遗传性的标准来区分。以器质性表皮痣为特征的症状有Schimmelpenning综合征、色素沉着性角化病、粉刺痣综合征、安哥拉毛痣综合征和Becker痣综合征。到目前为止,这些疾病的分子基础还没有确定。相比之下,以角质形成细胞痣为特征的综合征包括3种分子病因学已知的表型:Child综合征(先天性半侧发育不良伴鱼鳞状痣和肢体缺陷)综合征、2型节段性考登病和成纤维细胞生长因子受体3表皮痣综合征(García-Hafner-Happle综合征),而Proteus综合征的起源尚不清楚[5]。目前仍存在与表皮痣相关的一组较不明确的表型,包括毛囊痣综合征、营养不良再生障碍性皮肤病、头皮综合征(皮脂痣、中枢神经系统畸形、先天性再生障碍性贫血、角膜缘皮损)[6]。一般认为本病为散发性,少数病例为常染色体显性遗传[7],本例患者家族无类似患者。目前认为基因突变在该病中扮演着重要角色,少数病例具有分子特征,存在体细胞激活RAS、FGFR3和PIK3CA突变[8]。Igawa[9]发现1例表皮痣综合征患者出现新的合子后KRAS突变,表现为角质形成细胞表皮痣和皮脂腺痣两种不同的临床特征。
该病的伴随症状具有多样性。Ullah等[10]报道了首例伴有三联体中枢神经系统畸形的“表皮痣综合征”;Adams等[11]报告1例严重的表皮痣综合征,除皮肤受损外,还累及口腔、咽部和中枢神经系统;Menni等[12]报道1例累及口腔,伴有腭裂;Kumar等[13]报道1例炎性线状疣状表皮痣,累及骨骼系统。该病也可伴有罕见的广泛血管侵犯,以动脉狭窄、动脉瘤形成或血管阻塞为主[14]。本例患者有明显皮肤改变、智力低下、运动发育落后,合并难治性癫痫,皮损经过组织病理证实为疣状表皮痣,诊断为表皮痣综合征明确。本病应与黑棘皮病相关的多种遗传病鉴别,包括Alsrom综合征(视网膜病变、进行性感音神经性听力下降、向心性肥胖)、Crouzon综合征(面瘫、感音神经性听力下降、骨骼和智力发育迟滞)、Seip-Lawrence综合征(先天性脂肪营养不良性糖尿病)、Costello综合征(出生后生长缺陷、粗糙面容、累及颈部、掌跖和手指的皮赘、深色皮肤和乳头状瘤)、Bannayan Riley-Ruvalcaba综合征(皮下脂肪瘤、血管畸形、阴茎和女阴雀斑样痣、疣状皮损、大头畸形、智力低下、肠道息肉、骨骼异常、中枢神经系统血管畸形和甲状腺肿瘤)、Lelis综合征(外胚层发育不良、多毛、少汗、牙发育不全、掌跖角化过度和口周皱纹)[15]。
本病多数在出生即有或出生不久就有皮肤表现,随着年龄的增长出现其他系统表现,严重影响患者的外貌及生活质量,目前尚无特殊治疗。有研究对20例表皮痣患者进行Er∶YAG激光烧蚀治疗,患者有明显的临床改善,无任何不良反应,12个月内无复发[16]。皮肤损害可用液氮冷冻或激光治疗,应观察和处理中枢神经系统、心脏和肾脏的改变。
猜你喜欢
建材发展导向(2021年14期)2021-08-23 00:56:24
祝您健康·文摘版(2019年4期)2019-06-11 10:20:34
东方教育(2017年14期)2017-09-25 02:07:38
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18 10:59:54
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18 10:59:54
课程教育研究·学法教法研究(2016年1期)2016-03-17 15:52:58
中国房地产业(2016年9期)2016-03-01 01:26:33
西南医科大学学报(2015年1期)2015-08-22 13:02:00
中华皮肤科杂志(2014年4期)2014-12-19 12:55:46
浙江中西医结合杂志(2014年10期)2014-07-07 15:47:01
