
Preparation of a N, S, P co-doped and oxidized porous carbon for the efficient adsorption of uranium(VI)
2021-12-29 02:29LIUYanLIUXiaopengDAIYingWANGYunYUANDingzhongLIUJinbiaoCHEWJiawei
新型炭材料 2021年6期
LIU Yan*, LIU Xiao-peng DAI Ying WANG Yun YUAN Ding-zhongLIU Jin-biao, CHEW Jia-wei*
(1. State Key Laboratory of Nuclear Resources and Environment, East China University of Technology, Nanchang 330013, China;2. Department of Chemistry, Jiangxi University of Science and Technology, Ganzhou 341000, China;3. School of Chemical and Biomedical Engineering, Nanyang Technological University 637459, Singapore;4. Singapore Membrane Technology Center, Nanyang Environment and Water Research Institute,Nanyang Technological University 639798, Singapore)
Abstract: A N, S, P co-doped and oxidized porous carbon was prepared by the carbonization of poly (cyclotriphosphazene-co-4,4’-sulfonyldiphenol) at 750 °C, followed by KOH activation and HNO3 oxidation. The carbon was used as an adsorbent for uranium(VI) in aqueous solutions. TEM, SEM, XPS and FTIR were used to characterize its microstructure before and after adsorption.Results indicate that there is an optimum pH value of 6 for U(VI) adsorption. The adsorption kinetics and isotherms were fitted well by the pseudo-second-order and the Langmuir models, respectively. The maximum adsorption capacity determined by the Langmuir model at 298 K and a pH value of 6 was 402.9 mg g−1. The carbon has excellent reusability and retains 70% of the capacity of the original value after five adsorption-desorption cycles. The high U(VI) adsorption capacity is mainly attributed to the carboxyl, and P and S groups by the formation of the UO22+(COO−)2 complex, and U―O―P and U―O―S bonds.
Key words: Polyphosphazene;Carboxyl;Uranium;Adsorption;Heteroatoms
1 Introduction
Uranium is a radioactive pollutant and primitive heavy metal, which is harmful for the environment and public health[1]. Uranium is hazardous to living beings because its accumulation in organisms induces an increasing danger to global public health[2]. Therefore, it is urgent and necessary to have an effective approach to clean up uranium from aqueous solutions[3–5].
To date, a variety of approaches have been proposed to remove U(VI) from wastewater, including chemical precipitation, photocatalytic reduction,phytoremediation and adsorption[6–11]. Of these, the adsorption method has received substantial attention and has been proved to be an efficient method for removing uranium owing to its low-cost, versatility and simplicity[12].
During the practical use and in the development of effective adsorbents, the properties of adsorbents are crucial[13]. Numerous classes of materials have been investigated as sorbents, including graphene,mesoporous silica, and ion-imprinted magnetic chitosan resins[14–17]. Carbon-based materials are gaining increasing interest owing to their large surface area, and distinctive chemical and physical properties[18]. Carbon spheres (CSs), have been used as sorbents to remove pollutants because of their tunable sphere size, low surface energy and other advantageous characteristics[19]. Most synthesized CS surfaces show graphitic character and dangling bonds,which significantly improve the surface reactivity, favoring the utilization as a sorbent[20]. Many studies have been reported the fabrication methods of CSs, including pyrolysis of hydrocarbons, the hydrothermal method and polymerization followed by carbonization. Among these methods, polymerization has many merits in terms of the high porosity of the sorbents and the easy functionalization of the surfaces by grafting new reactive groups to improve their affinity or selectivity for target metals[21]. Phosphorus, nitrogen or sulfur-doped carbon materials are gaining increasing interests in the adsorption field. Incorporation of different heteroatoms into the sp2carbon structure has been proved to improve the adsorption properties of several carbon materials. Zhao et al.[22]reported a graphene sponge (GS) doped by S, which was synthesized by a hydrothermal method and had a Cu2+sorption capacity of 228 mg g−1, 40 times higher than that of activated carbons. Wang et al.[23]used N,P and S codoped graphene-like carbon nanosheets(NPS-GLCs) as adsorbents to adsorb uranyl ions.They found by XPS and DFT calculation that the adsorption mechanism was the strong covalent bonds between the heteroatoms with UO22+such as P-O-U and S-O-U. N, P or S-doped carbon materials have unique structural advantages with abundant adsorption sites incorporated in the carbon framework.
Polyphosphazenes can be formed by controlled and simple synthesis routes and have been proven to be ideal precursors of porous CSs due to the escape of a part of heteroatoms to form pores during carbonization, which is favorable to improve the adsorption capacity[24]. However, the majority of carbon materials are decorated with various functional groups to improve the affinity and selectivity towards uranyl ions.Generally, water containing radionuclide wastes is acidic and carbon materials with carboxyl-functional groups show higher adsorption capacity in acidic solutions than that with other conventional oxygen containing functional groups[25]. Unlike traditional adsorbents, the adsorption behavior and mechanism of the heteroatoms-doped carbon materials have not been adequately explored[26].
Hence, in this paper we report a method for the fabrication of a carboxyl-functionalized carbon material doped with N, P and S simply by pyrolysis of polyphosphazenes (denoted as CS-COOH). The resulting CS-COOH was characterized to analyze the chemical structure and used as a sorbent for removing Ufrom aqueous solutions using a series of batch experiments. The effect of pH, initial U(VI) concentration, contact time and recyclability were also assessed. This study illustrates that CS-COOH could adsorb U(VI) effectively and is promising for the applications in wastewater treatment.
2 Experimental
2.1 Synthesis of CS-COOH
The synthesis route of CS-COOH is illustrated in Fig. 1. Cross-linked PZS (poly(cyclotriphosphazeneco-4,4’-sulfonyldiphenol)) spheres were synthesized from hexachlorocyclotriphosphazene (HCCP) and(BPS) through in-situ polymerization[5]. Then, the PZS spheres were calcined at 750 °C at a heating rate of 4.5 °C min−1under N2atmosphere to produce the nitrogen, phosphorus and sulfur atoms doped carbon nanosheets[23]. After cooling down, the codoped carbon material was mixed with KOH (used as an activating agent) at a weight ratio of carbon to KOH of 1∶3, and then the mixture was heated in the furnace to 800 °C at a heating rate of 5 °C min−1and held for 1 h under N2atmosphere to carry out activation. The activated carbon material was washed with 10 wt%HCl until the solution pH value reached 7. Finally,CS-COOH was prepared by refluxing 100 mg of the activated carbon material in a 250 mL solution of 50%w/wHNO3at 140 °C for 2 h under stirring vigorously[27]. The resulting product was filtered, washed with distilled water and ethanol, and dried at 80 °C for 12 h under vacuum.
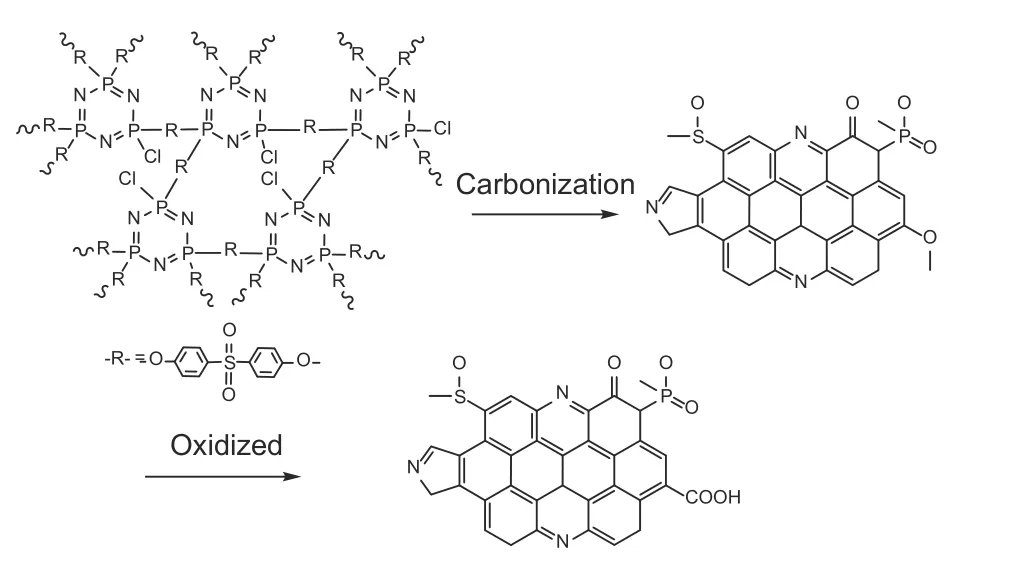
Fig. 1 Preparation process of CS-COOH.
2.2 Adsorption experiments
A uranium concentration of 1 mg mL−1was prepared by dissolving 2.110 g of UO2(NO3)2·6H2O into a mixture of hydrochloric acid (36.5 wt%) and hydrogen peroxide (36.5 wt%). The uranium solutions with different concentrations were prepared by diluting the original solution with deionized water.
Then the snow began to fall so heavily that the little boy could not see a hand’s breadth before him, but still they drove on; then he suddenly loosened the cord so that the large sled might go on without him, but it was of no use, his little carriage held fast, and away they went like the wind
For each experiment, 5 mg of CS―COOH was mixed in 30 mL of U(VI) solutions at different pH values, concentrations and temperatures. When the adsorption equilibrium was reached, the concentrations of the initial and final solutions were determined by the arsenazo-III UV-vis spectrophotometer. The amount of adsorbed (qe, mg g−1) uranium was calculated by Eq. (1):
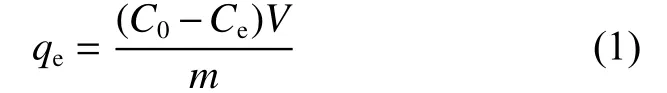
whereC0andCe(mg L−1) are the initial and equilibrium concentrations of U(VI) in the solutions, respectively,Vis the volume of the solution (L) andmis the mass of the dry adsorbents (g). Furthermore, the adsorption capability of the adsorbent was characterized by the distribution coefficientK:
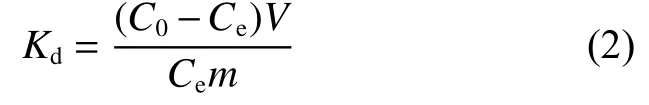
2.3 Characterization
The functional groups of the sample was analyzed using the FTIR (TENSOR27 FTIR spectroscopy (Bruker)) in the wavenumber range of 4 000 to 400 cm−1. Morphological analysis was carried out using a JEOL JSM-5900 SEM and a JEM-2100 TEM.XPS spectra were obtained on a kratos Axis Ultra DLD. Spectrophotometric investigation was measured by the UV-vis spectrophotometer (757CRT,Tianjin Guanze Technol. Co., Ltd). The high resolution transmission electron microscopy (HRTEM,America FEI) was conducted at an accelerating voltage of 300 kV. The specific area was determined based on the Brunauer-Emmett-Teller (BET) equation from the nitrogen adsorption isotherm on a Micromeritics Tristar (USA).
3 Results and discussion
3.1 Characterization
SEM and TEM images of CS and CS―COOH are presented in Fig. 2. The prepared CS spheres are highly uniform with an average size of about 300 nm without any aggregation (Fig. 2a and 2c). After oxidization, the CS spheres (Fig. 2a) were converted into porous CS―COOH that exhibits a rougher surface as compared with the CS spheres (Fig. 2b). The TEM and HRTEM images (Fig. 2d-f) show that the CS―COOH exhibits a graphene-like structure and the layers of carbon nanosheets are stacked together.
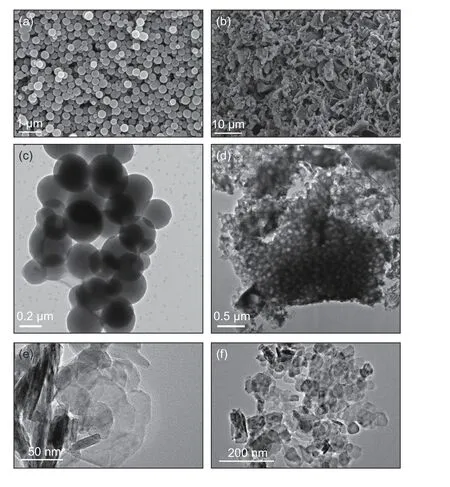
Fig. 2 SEM, TEM and HRTEM images of (a, c) CS and(b, d-f) CS-COOH.
The porous structures of CS and CS―COOH were further investigated by N2adsorption in Fig. 3.The curves show a type IV isotherm with a typical H3 hysteresis loop. The BET specific surface areas of CS and CS―COOH are calculated to be 70.31 and 103.48 m2g−1, and the corresponding pore sizes are 36.21 and 20.37 nm, respectively.
The FTIR patterns of CS and CS―COOH are given in Fig. 4a. The peak at 1 500 – 1 600 cm−1indicates the existence of carbon structures and C=N,which confirm the existence of doped N. The peak at 990 cm−1corresponds to the stretching vibration of Ar-O-P, which can be attributed to the condensation between HCCP and BPS. The peaks at 3 415, 1 263 and 1 407 cm−1are ascribed to the stretching vibration of-OH, S-O and C-N, respectively. Compared with the spectrum of CS, the FTIR spectrum of CS―COOH shows a new absorbance peak at 1 718 cm−1, which suggests the formation of C=O after oxidization of CS.
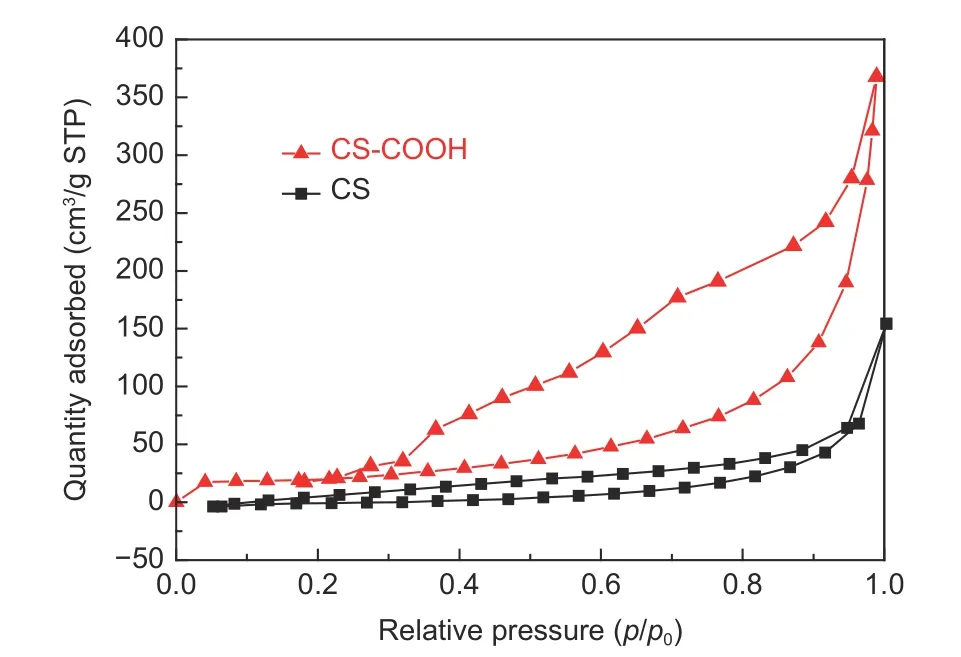
Fig. 3 N2 adsorption-desorption isotherms of CS and CS―COOH.
The XPS analysis was conducted to evaluate the chemical structures of CS―COOH and CS. As shown in Fig. 4b, the peaks of CS―COOH at 284.8, 400.3,163.9, 532 and 133.3 eV are attributed to C1s, N1s,S2p, O1s and P2p, respectively. The oxidizing reaction results in the increase of O-containing groups of CS―COOH, so the intensity of the O1s peak of CS―COOH is stronger than that of CS. The peak intensity of oxygen-related functional groups (-COOH)agrees with the FTIR results.
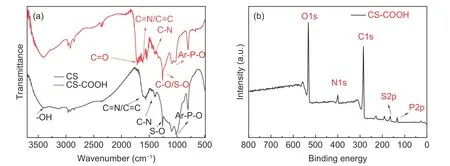
Fig. 4 (a) FTIR spectra of CS and CS―COOH and (b) high resolution XPS spectrum of CS―COOH.
3.2 Effect of the initial solution pH value
In general, the influence of the pH value on the sorption capacity is mainly determined by the balance between attractive forces and repulsive forces[29]. The carboxyl groups are difficult to be deprotonated at low pH values and therefore are incapable to coordinate with UO22+. At the pH value range of 5-6, the carboxyl groups are deprotonated and coordinate with UO22+,forming a UO22+(COO−)2complex[17]. Fig. 5b shows the distribution of U(VI) species at various pH values.At the pH values of 5-10, U(VI) are negatively charged in the form of UO2(OH)42−[30], which are unfavorable for adsorption due to the repulsive interaction between the nagatively charged U(VI) species and the deprotonated-OH and-COOH groups on CS and CS―COOH.
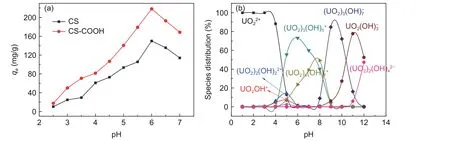
Fig. 5 (a) Effect of initial solution pH on the adsorption of U(VI) by CS and CS―COOH. (T = 298 K, m = 5 mg, C0 = 50 mg L−1 and V = 30 mL);(b) Distribution of U(VI) species in aqueous solutions with a total concentration of 50 mg L−1 and pH values ranging from 1 to 12 (calculated by using a Medusa program).
3.3 Adsorption kinetics
The influence of contact time on adsorption efficiency of CS and CS―COOH on U(VI) at 298 K was investigated (Fig. 6a). The adsorption capacities of CS and CS―COOH reach equilibrium after 80 and 100 min, respectively. The quick adsorption in the initial absorptive stage within about 40 min is mainly attributed to the availability of a large number of adsorption sites and the strong binding of uranyl ions with functional groups.
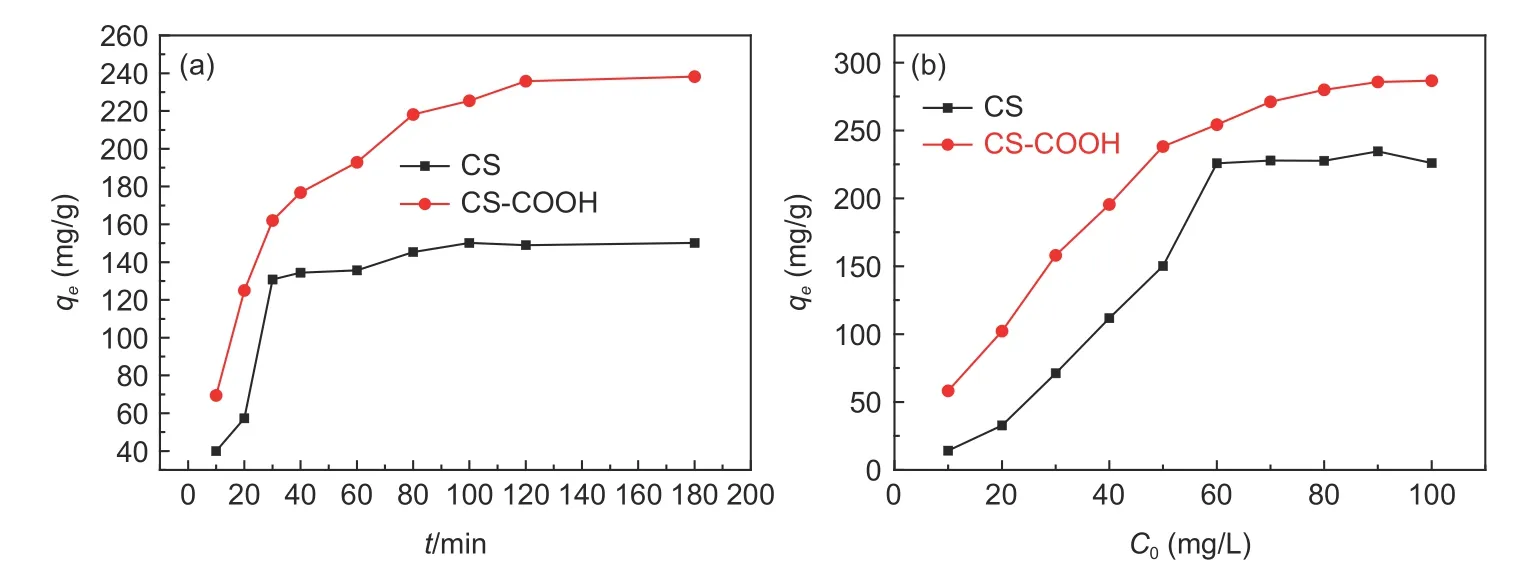
Fig. 6 (a) Effect of (a) the contact time (C0 = 50 mg L-1) and (b) the initial concentration on the adsorption of U(VI) by CS and CS-COOH.(T = 298 K, m = 5 mg, V = 30 mL and pH = 6)
The pesudo-first and -second order models were applied to understand the sorption process of U(VI) on CS and CS―COOH. The specific formulas of the two models are shown in Eq. 3 and 4. The non-linear fitting curves results are presented in Fig. 7 and the model parameters and the correlation coefficients are listed in Table 1.
Pseudo-first-order model:
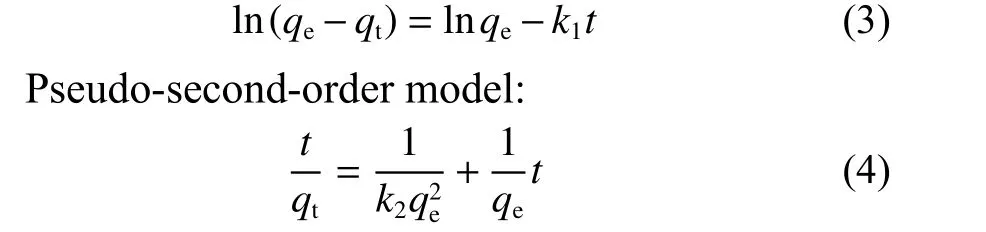
whereqeandqt(mg g−1) are the adsorption capacities at equilibrium and at timet(min), respectively,k1andk2are the rate constants for the pseudo-first-order (min−1) and pseudo-second-order models(mg−1·min−1), respectively.

Fig. 7 (a) Pseudo-first-order and (b) pseudo-second-order adsorption kinetics models of U(VI) adsorption by CS and CS―COOH.

Table 1 Kinetic parameters for adsorption of U(VI) by CS and CS-COOH.
The correlation coefficients of the pseudosecond-order model are higher than 0.97 for CS and CS―COOH. The calculated adsorption capacities(q2,cal) of 173.31 and 250.43 mg g−1for CS and CS―COOH, respectively, are close to the experimental adsorption capacities of 150.34 and 238.20 mg g−1for CS and CS―COOH, respectively. This suggests that the rate-controlling step might be chemisorption rather than physical sorption. Chemisorption may be the valence force involved in interchanging or sharing electrons between U(VI) ions and adsorbents[6].
3.4 Adsorption isotherms

Fig. 8 (a) Langmuir and (b) Freundlich isotherms of U(VI) adsorbed by CS and CS-COOH.
The relationship between the initial concentration and adsorption capacity is essential to figure out the process of U(VI) adsorption[31]. Fig. 6b displays the influence of the initial concentration on adsorption capacities of CS and CS―COOH to U(VI) at a pH value of 6. It is found that the adsorption capacities of CS and CS―COOH increase with increasing the initial concentration of U(VI), and the highest sorption capacities reach 234 and 286 mg g−1, respectively.
The Langmuir (Eq. 5) and Frenudlich (Eq. 6)models are used to fit the curves in Fig. 8 and the corresponding parameters are listed in Table 2. From the correlation coefficients (R2), it can be concluded that the Langmuir model is better than the Freundlich model in the fitting of U(VI) adsorption data on CS and CS―COOH, implying that the active sites of CS and CS―COOH are covered by U(VI) with a mono-layer mode. The maximum adsorption amount calculated by the Langmuir model is 229.36 mg g−1for CS and 402.90 mg g−1for CS-COOH at 298 K.
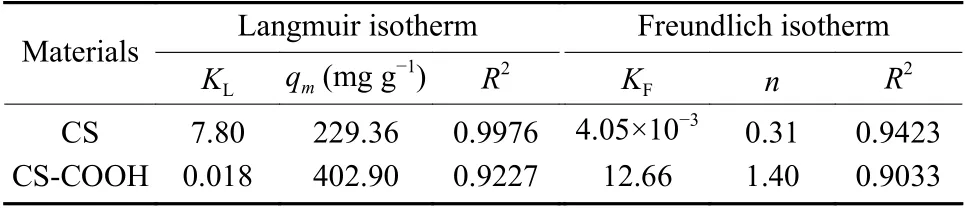
Table 2 Parameters of Langmuir and Freundlich isotherm models for adsorption of U(VI) by CS and CSCOOH.
Langmuir model:
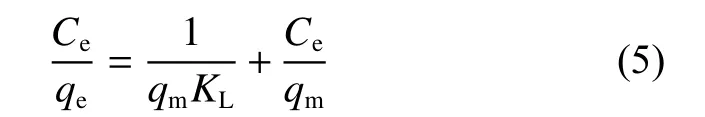
Frenudlich model:
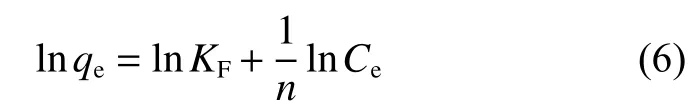
whereqeis the equilibrium adsorption capacity(mg g−1),qmstands for the maximum capacity (mg g−1),KLis the Langmuir adsorption equilibrium constant(L mg−1),Ceis the concentration at equilibrium(mg L−1), andKFandnare the Freundlich constants representing adsorption capacity and intensity, respectively.
3.5 Thermodynamic parameters
In order to confirm the influence of the temperature on adsorption behavior, the adsorption capacities of CS and CS―COOH on U(VI) are shown in at various temperatures under similar experimental conditions are shown in Fig. 9a[21]. The thermodynamic parameters ΔG, ΔSand ΔHfor the adsorption of U(VI) are listed as follows:
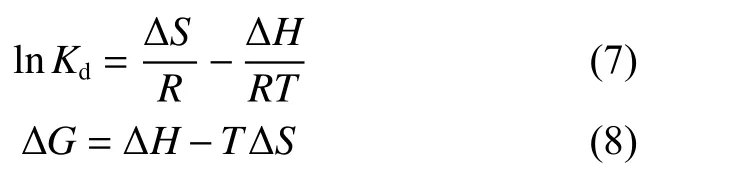
where ΔH(J mol−1), ΔS(J mol−1K−1) and ΔG(J mol−1) are the enthalpy, entropy and Gibbs free energy, respectively.

Fig. 9 (a) Effect of the temperature on the equilibrium adsorption capacities of U(VI) on CA and CS―COOH (m = 5 mg, V = 30 mL, C0 = 50 mg L−1,t=180 min and pH = 6) and (b) the equilibrium constants as a function of 1/T of U(VI) adsorption by CS and CS―COOH.

Table 3 The thermodynamic parameters for U(VI) adsorption by CS and CS-COOH.
As shown in Table 3 and Fig. 9b, the negative values of ΔGat different experimental temperatures indicate the spontaneous nature of the adsorption process, while the negative values of ΔHsuggest the exothermic nature of the adsorption process[32]. The positive values of ΔSimply an increase in the disorderliness of the solid-solution system[29].
3.6 Recyclability and the effect of competitive ions
The selectivity of CS and CS-COOH for U(VI)adsorption was evaluated in the presence of competing metal ions (Co2+, CS+, Na+, Zn2+, Ni2+and Mn2+),with the equimolar U(VI) (0.5 mmol L−1) (Fig. 10a).As evidenced, the CS―COOH exhibits stronger adsorption abilities to U(VI), indicating its high affinity and selectivity for U(VI).
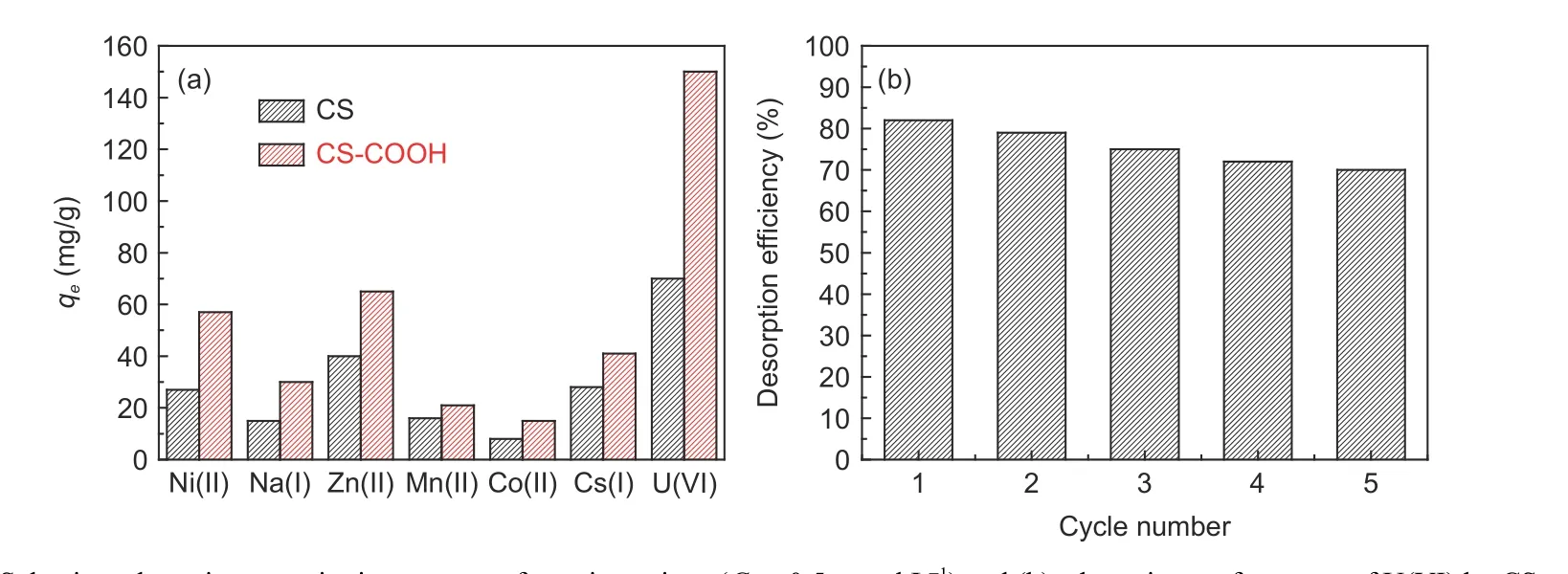
Fig. 10 (a) Selective adsorption capacity in presence of coexistent ions (C0 = 0.5 mmol L−1) and (b) adsorption performance of U(VI) by CS―COOH over 5 cycles (C0 = 50 mg L−1). (m = 5 mg, V = 30 mL, T = 298 K and pH = 6).
An excellent reusability of a adsorbent is important to reduce the expense in the practical application[31]. Thus, the recycling of CS―COOH was investigated over 5 continuous cycles of sorption/desorption and the efficiency of each cycle is presented in Fig. 10b. It is observed that UO22+can be eluted with 0.5 mol L−1HCl solution. It is clear that, after 5 cycles, CS―COOH retains about 70% of the original adsorption capacity. The decrease may be attributed to a part loss of oxygen-containing functional groups on CS―COOH during the elution process. The findings affirm that CS―COOH is a promising adsorbent for uranium solution clean-up in practical applications.
3.7 Mechanism analysis of adsorption
XPS analysis was employed to investigate the interaction between U(VI) species and CS―COOH.The XPS spectrum of CS―COOH loaded with U(VI)(denoted as CS―COOH―U) is shown in Fig. 11a.The CS―COOH―U displays characteristic peaks at 284.8, 531.8, 400.3, 133.2 and 163.9 eV that corresponds to C1s, O1s, N1s, P2p and S2p, respectively,while double peaks of U 4f appear at 393.0 and 382.3 eV that are associated with U4f5/2and U4f7/2, respectively. The location of U4f7/2is shifted rightwards with respect to the standard reference datum(382.45 ± 0.05 eV)[33–34], which can be attributed to the reduction of Lewis basicity due to the conversion from Uto U―O―C or U―O―P bonds[32,35].The high-resolution C1s spectra before and after adsorption are shown in Fig. 11b. The assignments of the relevant binding energies before adsorption for C 1s are as follows: 284.57 eV for C―C/C=C bonding (graphitic carbon), 285.37 eV for C―P/C―N bonding (phenolic groups) and 288.51 eV for C=O bonding (carboxyl groups). The O1 of CS―COOH before adsorption can be divided into three peaks(Fig. 11c), including 531.2, 532.27 and 533.18 eV for C=O, P―O/C―O―C and ―OH, respectively. The rightward shift of the P―O/C―O―C (531.33 eV)peak after adsorption is contributed by the combination of U(VI) with the carboxyl groups.
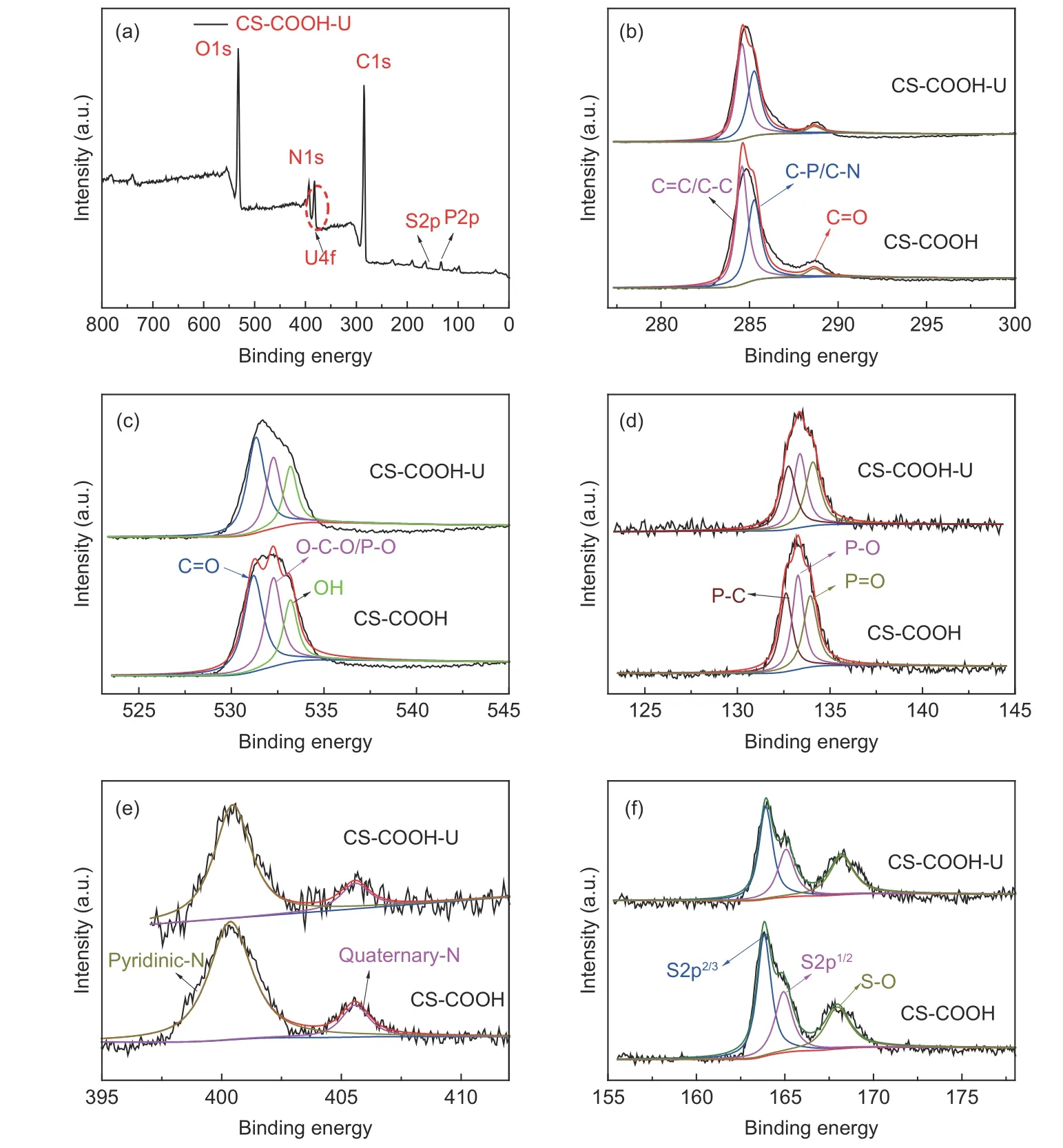
Fig. 11 (a) XPS survey spectrum of CS-COOH-U and the high resolution XPS spectra of (b) C1s, (c) O1s, (d) N1s, (e) P2p and (f) S2p.
As for the P2p spectra in Fig. 11d (P―C at 132.62 eV, P―O at 133.27 eV and P=O at 133.94 eV), the peak intensities change significantly after adsorption, which may be ascribed to the formation of U―O―P. In the N1s spectra of Fig. 11e, 2 peaks are found at 400.77 and 405.58 eV, corresponding to pyridinic-N and quaternary-N[36]. After adsorption, the 2 peaks remain unchanged. The S2p spectra(Fig. 11f) are deconvoluated into 2p3/2at 163.82 eV,2p1/2at 164.92 eV and S―O at 167.97 eV, while S―O (168.20 eV) shifts to a higher binding energy after adsorption, indicating the formation of U―O―S groups in U(VI) adsorption. The above results reveal that the carboxyl sites and the doped heteratoms in the CS―COOH contribute significantly to the adsorption capacity towards U(VI).
Besides, FTIR spectroscopy was used to investigate the adsorption mechanism of CS―COOH. A new peak of CS―COOH―U at 926 cm−1emerges in Fig. 12, which is attributed to O=U=O. Furthermore, the intensities of peaks corresponding to C―O/S―O decrease after adsorption, indicating chemical bonding between U(VI) with carboxyl groups and the doped S atoms.
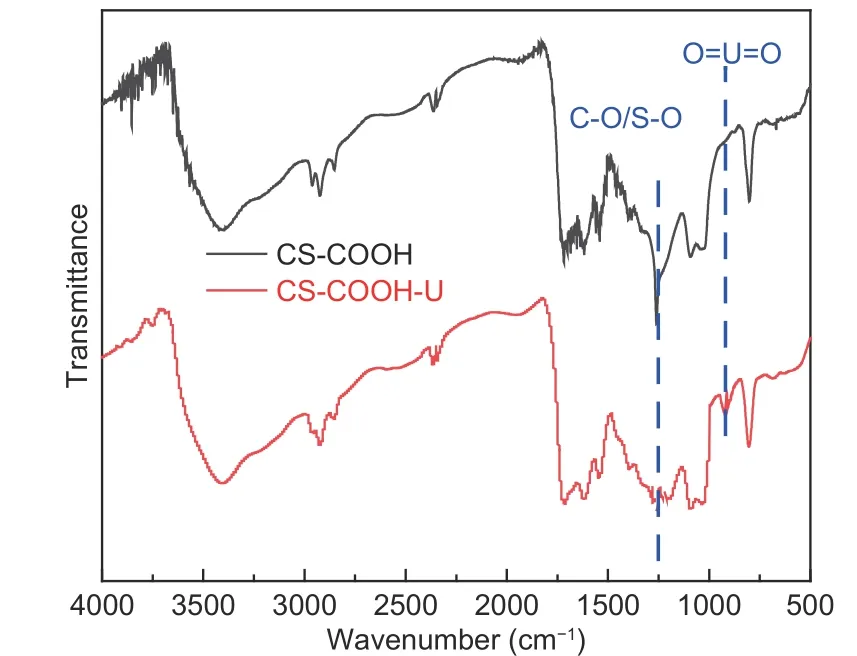
Fig. 12 FTIR spectra of CS-COOH and CS-COOH-U.
4 Conclusion
Herein, the N, P, S codoped carbonaceous spheres (CS) were prepared by carbonization of a polymer from a polyphosphazene precursor, which were activated with KOH and oxidized by HNO3to form CS―COOH. The removal of U(VI) was significantly influenced by the pH value with an optimal pH value of 6. The CS―COOH shows fast removal kinetics and a relatively high adsorption capacity(402.90 mg g−1at pH=6 andT=298 K). The thermodynamic analysis shows that the sorption process is endothermic. The effect of competing ions on the removal of U(VI) was also evaluated, which indicates that the selectivity of the adsorbent is high. Moreover,the XPS analysis also illustrates that the introducing of heteratoms changes the sorption process and provides more active sites. In conclusion,CS―COOH is an effective and promising adsorbent for the disposal of uranium-loading effluents.
Acknowledgements
We appreciate the financial support from the National Natural Science Foundation of China(No.22166001, 21966005, 22066002 ), the Jiangxi Provincial Natural Science Foundation (No.20202 BABL203001, 20192BAB202007, 20192ACB21001),the Opening fund project of State Key Laboratory of Nuclear Resources and Environment, East China University of Technology (NRE1926), Provincial College Students' Innovation and Entrepreneurship Training Program (S202110405019).

- 新型炭材料的其它文章
- 基于碳化钽涂层改性碳基材料的研究进展
- Two-dimensional layer materials for highly efficient molecular sensing based on surface-enhanced Raman scattering
- Coating a Na3V2(PO4)3 cathode material with carbon to improve its sodium storage
- Preparation of a N-P co-doped waste cotton fabric-based activated carbon for supercapacitor electrodes
- High-surface-area porous carbons produced by the mild KOH activation of a chitosan hydrochar and their CO2 capture
- Preparation of a porous carbon from Enteromorpha prolifera with excellent electrochemical properties