
High-surface-area porous carbons produced by the mild KOH activation of a chitosan hydrochar and their CO2 capture
2021-12-29 02:32:08WANGJingCHENShuangXUJiayuLIULichengZHOUJichengCAIJinjun
新型炭材料 2021年6期
WANG Jing, CHEN Shuang, XU Jia-yu, LIU Li-cheng, ZHOU Ji-cheng,*, CAI Jin-jun,*
(1. Hunan Key Laboratory of Environment Friendly Chemical Process, School of Chemical Engineering, Xiangtan University, Xiangtan 411105, China;2. CAS Key Laboratory of Bio-based Materials, Qingdao Institute of Bioenergy and Bioprocess Technology,Chinese Academy of Sciences, Qingdao 266101, China)
Abstract: Hydrothermal treatment of biomass is effective in producing hydrochar, but the product usually has a low surface area and is not suitable for direct use as an adsorbent for CO2 capture. We report the use of chitosan as a precursor for carbon prepared by a combination of hydrothermal treatment and mild KOH activation. The effect of an additive salt (eutectic salt of KCl/LiCl with a mass ratio of 5.5/4.5) in the hydrothermal treatment and activation temperature on the porosities and surface chemical states of the obtained carbons and their CO2 capture were studied by N2 adsorption, XPS, SEM and XRD. Results indicated that the porosities of the carbons were increased by increasing the activation temperature. The salt additive introduced mesopores in the hydrochar and slightly reduced the surface area of the porous carbon after activation, but was useful in increasing the number of N-species during hydrothermal treatment and activation. The carbons produced using the salt additive had much larger CO2 uptakes under ambient conditions than those prepared without the salt, suggesting that porosity is not the only factor that determines the CO2 uptake. The CO2 uptake on the carbon activated by KOH at 600 °C produced from the salt-assisted hydrochar was the highest (as high as 4.41 mmol/g) although its surface area was only 1 249 m2/g, indicating that CO2 uptake was determined by both the microporosity and the active N-species in the carbon.
Key words: Chitosan;Porous carbon;Hydrothermal carbonization;KOH activation;CO2 capture
1 Introduction
Accumulation of atmospheric CO2emitted from fossil fuels proposes urgent demand to capture CO2in a cost-effective way[1–3]. Liquid amine scrubbing is widely used to capture CO2by acid-base interaction while the regeneration needs energy consumption associated with amine volatilization and also there is a corrosion problem[4]. As an alternative measure, adsorption by solid porous materials such as zeolite,metal-organic frameworks, covalent-organic frameworks and porous carbon has been widely explored for CO2capture with low energy consumption[5].Among them, porous carbon is one of the most utilized ones owing to their well-developed porosity, lowcost, and chemical stability[6]. The micropores less than 1 nm play an important role in the CO2uptake at atmospheric pressure, whereas the micropores in the range of 1-2 nm as well as the mesopores have a positive effect at higher pressures[7–9]. N doping in the porous carbon is an efficient strategy to boost the CO2capture capability due to the special electron donating effect of N-species[10]. The importance of N-species for CO2capture has been widely investigated by several groups[11,12], which is efficient to improve the CO2uptake and CO2/N2selectivity due to the enhanced interaction between carbon surface and CO2molecules.
Porous carbon is traditionally obtained from the carbonization of coal, organic polymers or biomass materials[13,14]. To develop the porosity of the carbon material, further activation is required. Chemical activation could enhance the porosity of the carbon material at low temperature and short holding time. Activation agents, such as KOH[3,8], K2CO3[15], KHCO3[16,17], Na-OH[18]and NaNH2[1]have been explored for the production of porous carbons, and KOH attracts most attention due to its efficiency in creating micropores.From a viewpoint of green chemistry, much attention has been focused on the use of carbohydrates or biomass wastes to prepare porous carbon by hydrothermal treatment. As-made hydrochar has unique attributes such as high density of O-species and low condensation extent, which is favorable for further activation. For instance, a lotus stem-derived carbon was obtained by hydrothermal treatment and KOH activation[19], which had a surface area of 2 893 m2/g and a CO2uptake of 3.85 mmol/g under ambient condition.As a comparison, the direct carbonization of hydrochar without activation process only made a carbon with a surface area of 500 m2/g[20]. Yanget al.[21]used a similar two-step method to obtain a carbon from waste camphor leaves, which had a surface area of 1 633 m2/g and a CO2uptake of 6.63 mmol/g at 0.4 MPa. CO2capture performance could be affected by surface N modification via post-treatment or in-situ doping[15], while the detailed effect of N atoms is still not unclear with some disputation. Fanet al.[6]reported a porous carbon from glucose and urea by hydrothermal treatment and KOH activation, which had a CO2uptake of 4.37 mmol/g at 25 °C and 0.1 MPa, and the CO2/N2selectivity was highly up to 35 due to narrow micropores and some N-species. Strawberry was also converted into a carbon with N-species inherited from the precursor[22], which possesses a surface area of 1 117 m2/g and a CO2uptake of 4.49 mmol/g. Both KOH activation and N doping on the surface of carbons contribute to an increase in CO2uptake because of the increased microporosity and basic attribute of the surface. The eutectic salts are also usually added as porogenic agents to produce carbon materials with high microporosity. For instance, Qiu's group reported porous carbons with the surface areas of 451–1 190 m2/g from a molten-salt method using glucose as the carbon source and melamine as the nitrogen source[23], in which eutectic salts of LiCl/KCl were used as porogenic agents. As-made carbons showed N-species of up to 24% (mass ratios) with amazing H2S uptake at 25 °C. To minimize the corrosion effect caused by chemical activation, the amount of activatorsi.e.KOH during the activation process should be controlled in a limited scope. It is critically important to make N-doped carbons with limited amount of KOH, that is, a mild KOH activation process.
Chitosan has a similar structure to cellulose except for the substitution of ―OH by ―NH2group,which has great potential as a precursor for the synthesis of N-doped carbon as compared with other polysaccharidei.e.glucose, sucrose, and fructose[23–25].Chitosan itself also exhibits high removal performance for pollutants from water[26], because of its hydrophilicity afforded by a large number of ―OH groups, a flexible polymer chain and high reactivity.Yogoet al.[27]reported a porous carbon from chitosan using K2CO3as an activator, which has a CO2uptake up to 4.9 mmol/g at 0.1 MPa due to the presence of Nspecies. Their results also indicated that the small size K+as an activator would be effective to increase microporosity, resulting in the increase of CO2uptake.Recently, hydrothermal treatment was accepted as a simple route to convert biomass wastes or carbohydrates into hydrochar[20], and the presence of salts in the reaction system was useful for introduction of mesopores and O-species on the surface of hydrochar[28], which was beneficial for carbons with developed pores after activation. Nabaiset al.[24]reported a hydrochar from chitosan with N-species up to 5.9%, which was activated by CO2, K2CO3and CaCO3to prepare porous carbons with surface areas of up to 1 095, 2 130, and 1 432 m2/g, respectively. In their work, chitosan was hydrothermally carbonized and subsequently activated with a mild KOH activation process with a hydrochar/KOH mass ratio of 1∶2 to obtain carbons with appreciable pores and enlarged micropore apertures. A higher activation temperature of 800 °C largely enhanced the porosity with a surface area up to 2 547 m2/g, while the presence of salts in the hydrothermal systems seemed to have a slightly adverse effect when the hydrochar was activated at 600 and 700 °C. The carbons had much larger uptake as compared to that of counterparts without a salt,i.e. CN-0-700 and CN-5-700 showed CO2uptake of 2.97 and 3.45 mmol/g at 25 °C and 0.1 MPa, respectively, even if the latter had a slightly smaller surface area of 1 944 m2/g. The CO2uptake on CN-5-600 from KOH activation at 600 °C with a salt in the hydrothermal system was even up to 4.41 mmol/g, due to a synergistic effect from high microporosity and some N-species. The low-cost in production with appreciable CO2uptake indicated that the chitosan hydrochar was potential precursor for carbons as CO2adsorbents.
2 Experimental
2.1 Chemicals
Chitosan with a deacetylation degree of 85% was provided by Aladdin Reagent Co., Ltd. (Shanghai,China). KOH was used as an activator and a HCl solution was used to remove impurities. The eutectic salt was formed by grinding KCl and LiCl with a KCl/LiCl mass ratio of 5.5/4.5, according to the previous report[29]. Distilled water used in the whole experiments was prepared by a Millipore-Q water system in the lab.
2.2 Synthesis of chitosan-based porous carbons
Hydrothermal treatment was used to transform chitosan into hydrochar, where water of 60 mL with 10% (mass fraction) chitosan was held in a 100 mL autoclave at 180 °C for 12 h, and the eutectic salt was added into the systems as a catalyst or porogenic agent. As-made hydrochar was washed two times using hot water (80 °C) to remove the salt, dried at 100 °C, carbonized at 300 °C for 4 h, and activated with KOH at 600-800 °C for 1 h with a KOH/hydrochar mass ratio of 2 under N2flow with a ramping rate of 2 °C/min. The products were washed with 2 mol L−1HCl for 3 h, followed by distilled water until a neutral pH value reached, and dried at 100 °C for 24 h. As-made carbons were named as AC-x-t, where‘x’ indicated a ratio of salt to chitosan and ‘t’ indicated activation temperature.
2.3 Characterization
Thermal behavior of hydrochar was studied by a thermo-analyzer (TGA/1600HT, Mettler TOLEDO)up to 900 °C under N2flow. The microstructure of porous carbons were evaluated by Raman spectroscopy (Renishaw- inVia), X-ray diffraction (XRD,Bruker D8), transmission electron microscopy (TEM,JEM 2010), and scanning electron microscopy (SEM,JSM-6610LV, JEOL). Functional groups on the surface of carbons were studied by Fourier Transform-Infrared Spectroscopy (FT-IR, Nicolet380), and the detailed surface-doping structure was analyzed by X-ray photoelectron spectroscopy (XPS, Escalab 250Xi). N2adsorption-desorption isotherms of carbons were performed at −196 ºC on an automatic volumetric sorption analyzer (Micromeritics TriStar II 3020) after vacuum degassing at 180 ºC for 12 h, and CO2isotherms were collected both at 0 and 25 °C. The specific surface areas of carbons were calculated by the Brunauer-Emmett-Teller (BET) method, and pore size distributions (PSD) were calculated from the adsorption branches of N2isotherms according to the density functional theory (DFT) assuming a slit-pore geometry.
3 Results and discussion
3.1 Thermogravimetric analysis of hydrochar
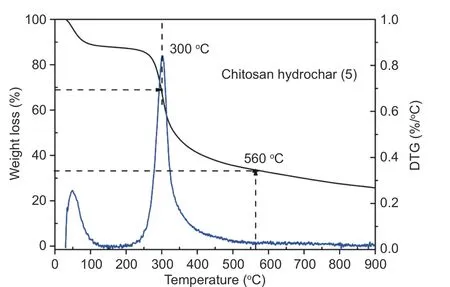
Fig. 1 TGA curves for chitosan hydrochar obtained with salt-assisted hydrothermal treatment under N2 atmosphere.
Thermal behavior of hydrochar obtained from salt-assisted hydrothermal treatment is shown in Fig. 1. Two distinct peaks were observed in the derivative thermogravimetric (DTG) curve, in which the former peak at 50 °C was attributed to moisture loss and the latter at 300 °C indicated the occurrence of phase transformation with the preliminary formation of carbon phases[30]. After that, no other evident peak was observed in DTG, and a further increase in temperature caused condensation of carbon atoms to form a highly porous network. TGA curve exhibited a distinct mass loss in the whole process. A distinct platform in the range of 50-250 °C indicated that the hydrochar was stable at this stage without impurities.The main loss stage from 250 to 560 °C with a mass loss of 54% was ascribed to the decomposition and volatilization of organic matter to form porous carbons. Most of unstable C atoms and noncarbon elementsi.e. H, N and O were removed as forms of small gas molecules, forming a network with solid residue enriched in carbon atoms[30]. The carbonization created porous tunnels in the bark hydrochar, the same as most of carbons from biomass materials[31,32]. A slightly mass loss beyond 600 °C was attributed to graphitization and elimination of functionalities. The carbon yield of chitosan hydrochar without activation was high with a remaining amount up to 33% at 800 °C.
3.2 Physical structure analysis for the materials
XRD patterns of different samples are shown in Fig. 2. A broad peak at 20° was observed for chitosan and hydrochar, indicating the attribution of crystalline polysaccharide in chitosan[23]. Hydrothermal would not destroy the crystalline structure. Other weak peaks at 10° and 15° were observed for hydrochar due to the formation of new phase in hydrolysis caused by automatically produced high-pressure environment. After activation, all peaks for hydrochar disappeared, indicating that mild activation was enough to wreck the partial crystalline structure of chitosan. XRD for all carbons showed two broad weak bands at 25° and 44°,ascribed to the (002) and (100) plane in typical carbons[30], respectively. The low intensity and broadness of peaks indicated the low graphitization degree,which were also observed for the carbons from the salt absent hydrothermal system. The peak at 25° was not observed for AC-5-800 while this peak was weak for AC-5-600 and AC-5-700, indicating that the severer activation would wreck the graphitic structure and enhance the porosity of carbons[8]. It has been noted that an empirical parameter (R) defined as a ratio of height of (002) Bragg peak to background as shown in Fig. 2 can be applied to estimate the number of single layer sheets in carbons[33]. In general,Rvalues are proportional to the content of parallel layers,that is, the largerRvalue meant the higher content of graphitic structure. The calculatedRvalues in AC-5-600, AC-5-700 and AC-5-800 were 1.7, 1.3 and 1.0,respectively. All results implied that KOH activation promoted the transformation of carbons into an amorphous structure with very limited graphitization.
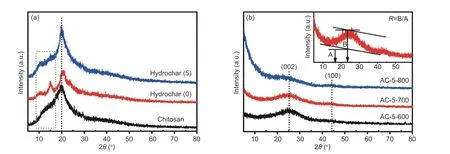
Fig. 2 XRD patterns: (a) chitosan and hydrochar, and (b) porous carbons after activation.
The morphological analysis in Fig. 3 revealed that hydrochar did not show a regular shape, similar to that of chitosan materials with wrinkles in the structure. The morphology of hydrochar was similar to chitosan-based carbons from direct carbonization, as already reported by Liet al[25]. It should be admitted that a spherical-like shape is a common attribute for hydrothermal process, while the reaction temperature and time largely affect morphology. Nabaiset al.[24]reported a chitosan hydrochar with a spherical-like shape obtained by carbonization at 200 °C for 24 h,while microspheres disappeared as temperature or time reduced to 180 °C or 12 h. Obviously, the present hydrothermal condition was inadequate to obtain hydrochar with a regular shape, and the thermal stability of chitosan rendered much higher resistance to hydrothermal treatment with very limited degradation of original structure. After activation, the morphology of carbons varied largely depending on activation conditions, and wrinkles in hydrochar were substituted by worn bulk particles with smooth surface as shown in Fig. 3b for AC-0-600. The morphology for AC-5-600 in Fig. 3c and AC-0-800 in Fig. 3d also had a similar shape as compared with AC-0-600. The higher temperature made the carbons with more destructive appearance due to the severer KOH intercalation effect, which was efficient for carbons with enhanced porosity. Fig. 3e and 3f show TEM images for AC-0-800, and the plenty of mesoporous cavities and micropores can be observed, indicating that hydrochar was fully corroded by KOH during activation.
3.3 Surface-doping chemistry of the materials
FT-IR spectroscopy was used to analyze functionalities of chitosan and carbons. It was observed from Fig. 4 that bandsi.e.1 432, 1 312, 1 261, 1 158,1 029 and 892 cm−1for chitosan disappeared after hydrochermal treatment and carbonization, indicating the depletion of heteroatomsi.e.O, H, and N in the formed hydrochar and carbons. Bandsi.e. 892 and 1 312 cm−1for chitosan were fully vanished in carbons, while they were still retained in hydrochar. A broad strong band at 3 400 cm−1was observed for all samples due to overlapping of stretching vibration from ―OH and ―NH2groups[25]. A number of Hbond chains usually existed in chitosan, and the chains with varied length and strength would cause stretching peak to form multiplets. The two peaks of 2 925 and 2 863 cm−1for chitosan and hydrochar were due to the stretching vibration from C―H groups[21], and they were fully vanished after activation with depletion of most H atoms from carbons. A broad strong band of 1 638 cm−1for the amide group was observed in chitosan[24], and the absence of splitting confirmed a high degree of deacetylation of up to 85% for chitosan as provided by the manufacturer. Significantly, the amide peaks still existed even after hydrothermal treatment and activation although their intensities were weakened, implying successful incorporation of N atoms into carbons. Two peaks at 1 381 &1 312 cm−1for the stretching of C―N/C=N groups were weakened after activation[24]. Considering the acid attribute of CO2molecules, basic sites associated with developed pores in carbons would pose a positive effect on the capture performance[12]. Moreover,the broad band with splitting peaks in range of 987–1 158 cm−1was ascribed to C―O or C=O bond stretching, where a peak at 1 080 cm−1was due to the vibration of glycoside[32]. Most of ether groups in hydrochar vanished after activation by the identification of C―O―C peak for anti-symmetric vibration at 1 261 & 1 158 cm−1and symmetric vibration at 1 029 cm−1. The band at 1 423 cm−1in chitosan was ascribed to the stretching of ―CH3which was weakened in the hydrochar and carbons due to the formation of =CH2groups from condensation reactions. All the FT-IR results indicated that hydrochar and carbons included N, O, and H atoms, and the numbers of oxygen and nitrogen-species in carbons were largely reduced after activation.

Fig. 3 Typical SEM images for different samples: (a) hydrochar (0), (b) AC-0-600, (c) AC-5-600, (d) AC-0-800 and (e, f) TEM images for AC-0-800.
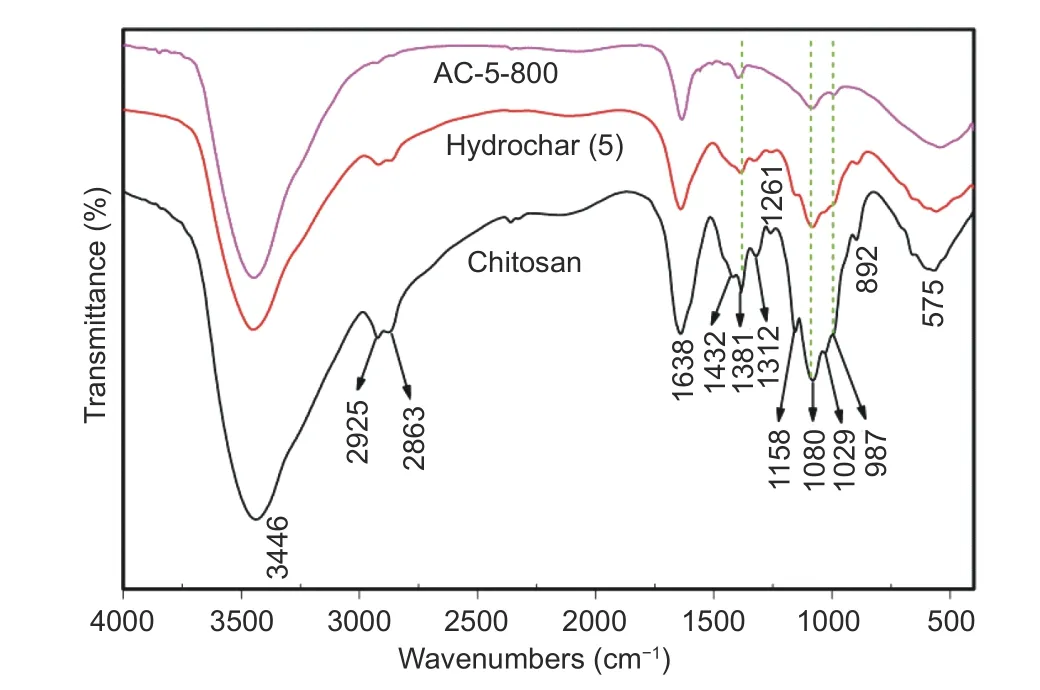
Fig. 4 Representative FT-IR spectra for chitosan materials,hydrochar and AC-5-800.
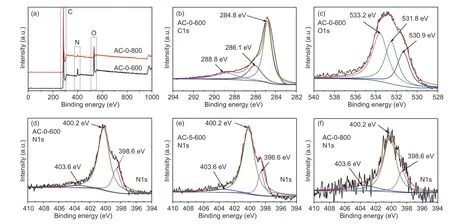
Fig. 5 XPS analysis: (a) Survey curves, (b) C1s and (c) O1s XPS spectra for AC-0-600 and N1s XPS spectra for(d) AC-0-600, (e) AC-5-600 and (f) AC-0-800.
The surface elemental compositions of three carbons were analyzed by XPS with results shown in Fig. 5 and Table 1. It was observed from survey spectra in Fig. 5 that AC-0-600 and AC-0-800 were confirmed to have C, N, and O atoms, and their peaks were found at binding energy of 284.8, 400.5 and 533.2 eV, respectively[34]. Other carbons also had similar results, indicating that chitosan was a suitable precursor for N-doped carbons with high doping levels because heteroatoms were not fully eliminated by carbonization and activation. Chemical compositions in Table 1 indicated that the content of C, O and N for AC-0-600 were 73.08 at.%, 20.19 at.%, and 6.73 at.%,respectively. Heteroatom contents were largely reduced as temperature went up to 800 °C and the contents of C, O, and N for AC-0-800 were 90.3%, 7.3%and 2.4%, respectively. The enriched C atoms and depleted O and N contents were caused by heteroatoms evaporation, and KOH also reacted with C, N, and O to form small gas moleculesi.e.H2O, CO2, CO, and NOxdue to the occurrence of dehydration, denitrogenation and decarboxylation[3]. High-resolution of C1s and O1s spectra in Fig. 5 for AC-0-600 indicated that the C1s spectra were deconvoluted into three peaks at 284.8, 286.1, and 288.8 eV, due to C=C,C=N/CHx/C―O, and O―C=O bonding, respectively[34,35]. The results for O1s XPS implied the presence of three types of O-species in the forms of COOH/C=O (530.9 eV), O―C=O (531.8 eV), and C―OH (533.2 eV)[3], indicating lactone, carboxyl, alcoholic and quinine groups existed in carbons. Demiret al.[35]reported lignin-derived carbons from a similar two-step route with the core peak at 531.2 eV for O1s spectra, while the present chitosan-based carbons had an obvious broad band (Fig. 5), indicating the precursor would largely affect the chemical state of carbons. The deconvolution of C1s and O1s XPS spectra in other carbons also showed similar results, indicating that the synthetic condition affected the contents of heteroatoms but did not change the type of chemical bonds. Bonding states of the N-species at 398.5,400.5 and 402.7 eV in Fig. 5 were attributed to three dominant configurations of pyridinic, pyrrolic, and graphitic-N, respectively[1,12]. The content of heteroatoms and the corresponding ratios of three N-species were summarized in Table 1, where N content decreased with an increase of temperature due to the more intensive etching procedures, and salt was efficient for consolidating N-species in carbons. Ratios for different N-species indicated that chemical states of N-doping can be changed by activation temperature. As a result, all the carbons showed the highest ratio of pyrrolic-N due to its high thermal stability with partial conversion from other type of N-species[30],which was believed to be favorable for CO2capture.

Table 1 Elemental contents of carbons determined by XPS analysis with the ratios of different N-species on the surface of carbons.
3.4 Porous structure analysis for materials
In general, pore generation caused by etching to produce small gas molecules was believed to be a main mechanism. N2isotherms and pore size distributions (PSD) of carbons are shown in Fig. 6. The detailed physical parametersi.e.surface area and volumes are listed in Table 2. As shown in Fig. 6, all N2isotherms had a typical type-I curve[11], and a large number of N2molecules were adsorbed atp/p0<0.02.All isotherms had little hysteresis loop in a whole pressure, indicating that carbons were essentially microporous. An almost flat feature was observed in all the isotherms with a limited increase in uptake asp/p0close to 1, where isotherms for carbons activated at 800 °C showed a wide ‘knee’ curvature, indicating the presence of small mesopores or macropores[1,8]. Multimode pore sizes were confirmed by PSD as shown in Fig. 6, where most pores were less than 5.0 nm and a small percentage of pores in the 20–80 nm range also existed. The wider ‘knee’ for AC-0-800 and AC-5-800 indicated the wider PSDs due to the more extensive etching. The smoother isotherms indicated higher microporosity[7,8]. All carbons showed well-developed pores with surface areas and pore volumes located in the range of 1 249– 2 547 m2/g and 0.67–1.39 cm3/g. Chitosan hydrochar was a suitable carbon precursor, and this mild activation with twofolds of KOH mass to hydrochar was effective to develop micropores. Both AC-0-800 and AC-5-800 had a lower microporosity due to the more extensive etching as compared with that of other carbons with the lower surface area (Table 2), indicating that the higher temperature was unfavorable for smaller micropores due to the partial collapse to form mesopores or even macropores. This statement was consistent to results of SEM images in Fig. 3. As compared with carbons without salt assistance, a limited enlargement in PSDs was observed for the carbons from hydrochar with salt presence in the hydrothermal systems. Considering a water-insoluble attribute of chitosan, salt was difficult to diffuse into inner structure of hydrochar, posing little effect on the expansion of pore aperture.
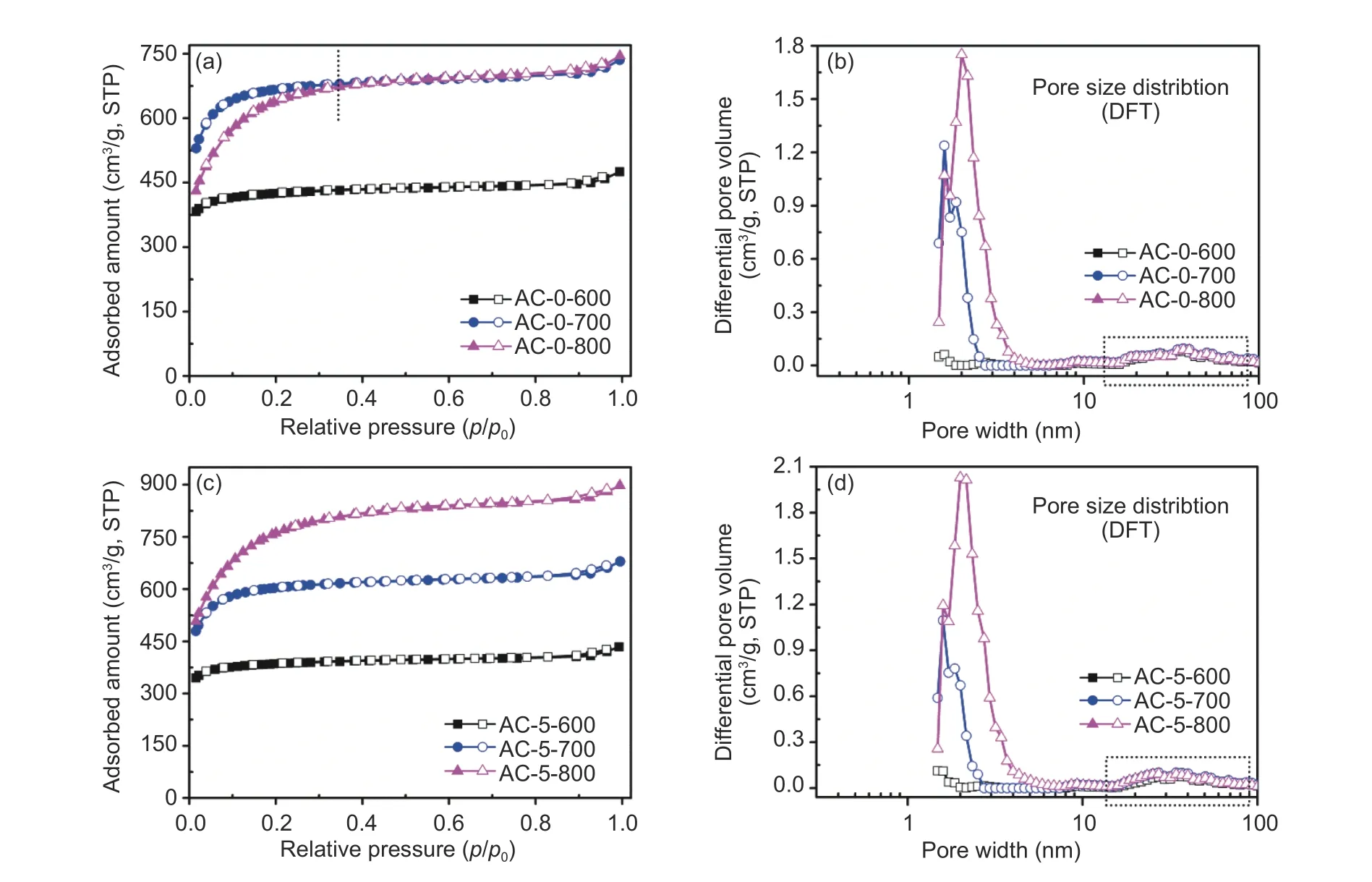
Fig. 6 Nitrogen adsorption-desorption isotherms for chitosan-derived carbons and corresponding pore size distributions determined by DFT model.
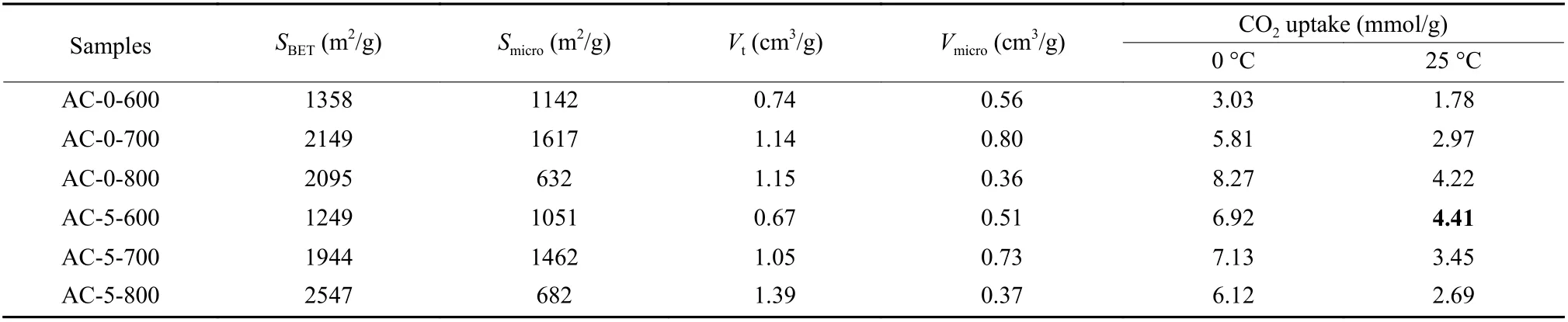
Table 2 Porous structure parameters and CO2 uptakes for the carbons from chitosan hydrochar.
3.5 CO2 uptake analysis for the materials

Fig. 7 The CO2 capture performance at (a) 0 °C and (b) 25 °C for chitosan-derived carbons.
The CO2capture performance was studied as shown in Fig. 7 with detailed uptakes at 0.1 MPa summarized in Table 2. As listed in Table 2, the uptakes at 0 °C on carbons were in the 3.03–8.27 mmol/g range and uptakes at 25 °C were in the 1.78–4.41 mmol/g range, which largely depended on surface areas of carbons but not soley proportional to surface areas and pore volumes[9]. The uptakes in Table 2 were highly reduced as temperature increased, indicating a physisorption mechanism for CO2capture. However, the CO2uptake was not proportional to the temperature.AC-0-800 with a surface area of 2 095 m2/g showed the largest uptake of 8.27 mmol/g at 0 °C, while the uptake at 25 °C was not the largest among series of carbons with the value of 4.22 mmol/g. The largest uptake of 4.41 mmol/g at 25 °C was observed for AC-5-600 with a surface area of 1 249 m2/g. The overall results indicated that the highest CO2uptake for chitosan-based carbons was contributed by a combination of porosity from smaller micropores and suitable N-species[9,36]. Isotherms showed an increase trend of uptake with increasing pressure in Fig. 7, and no evident saturation was observed at 0.1 MPa. The higher uptake would be obtained if pressure was further increased, especially for AC-0-800 with a much steeper increase trend in uptake, considering an enlarged PSD from the severer etching at higher temperature[8]. AC-5-800 with the largest surface area did not show the largest uptake, indicating that the total porosity was not a single factor in determining the CO2uptake[9].AC-5-600 with a surface area of 1 249 m2/g had the largest uptake at 25 °C and 0.1 MPa, among the highest level as compared with other types of biomass-derived carbons with large surface areas. Actually, the uptakes on AC-5-600 had only less than 1%loss even after five consecutive cycles (not shown here), indicating the amazing cycle and stability performance of chitosan-derived carbons as the adsorbents for CO2capture. All the collected data in Table 3 indicated that porous carbons activated at mild conditions with more small-sized micropores and N-species had high CO2uptakes under ambient condition. It was not hard to understand phenomena from N2isotherms in Fig. 6 and XPS spectra in Fig. 5, AC-5-600 exhibited the highest pyrrolic-N and microporosity especially for pores from narrower micropores due to an occurrence of activation at lower temperature[15,30,32].As observed from Table 2, the total pore volume for AC-5-600 was 0.67 cm3/g, while the corresponding micropore volume was highly up to 0.51 cm3/g, show-ing the highest microporosity. AC-0-600 without salt had much higher microporosity, while uptake at 25 °C was only 1.78 mmol/g due to its low surface area. The results confirmed the importance of N-species on the CO2uptake especially from pyrrolic-N[31], due to a consolidation effect of N-species from salt. Therefore,both the ratios of microporosity especially for porosity from the small-sized micropores and the number of active N-species contributed positive effect on CO2capture performance.
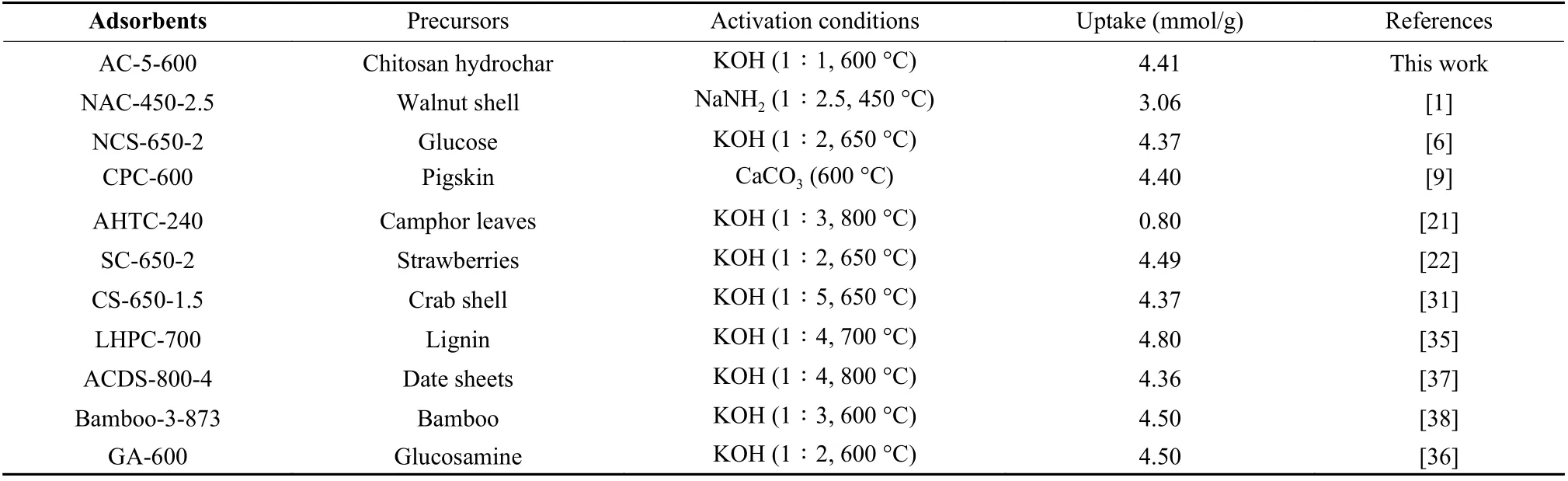
Table 3 CO2 uptakes (mmol/g) on various types of biomass-derived carbons at 25 °C and 0.1 MPa.
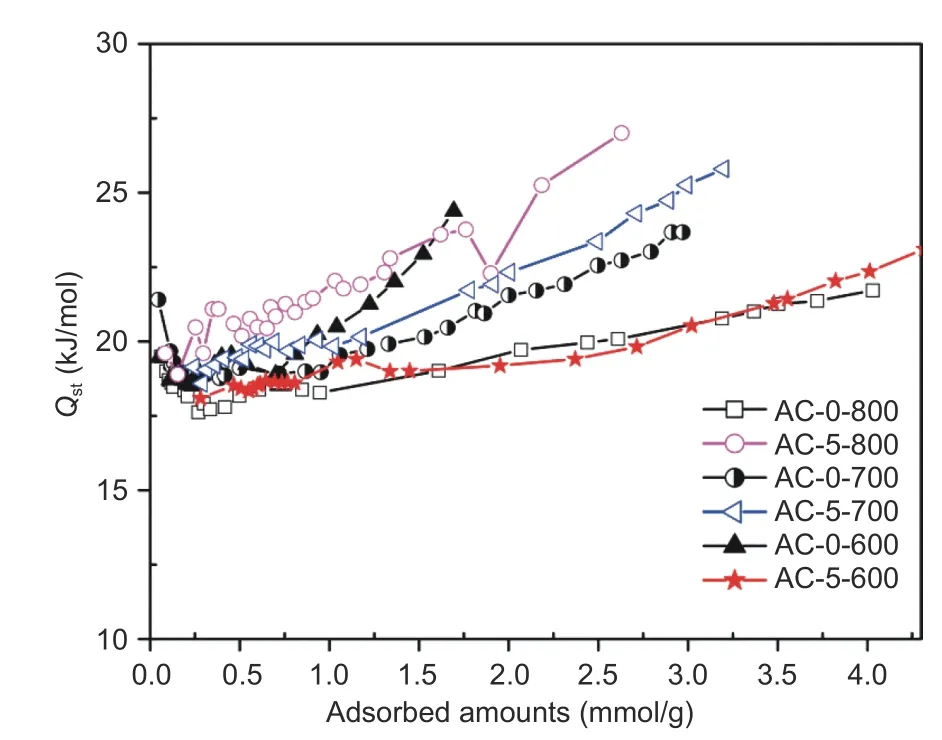
Fig. 8 Isosteric heat (Qst) of CO2 on the surface of carbons.
To better understand the interactions between CO2molecules and adsorbents, isosteric heat (Qst) of CO2on carbons were calculated from the isotherms at 0 and 25 °C using the classical Clausius-Clapeyron method[11,19]. As illustrated in Fig. 8, allQstvalues were in the range of 18–27 kJ/mol, smaller than that of other types of biomass- derived carbons especially for carbons with attributes of high surface areas, high microporosity and moderate N-species[1,19,32], suggesting that physisorption dominated CO2capture performance. In general, initialQstvalue was the largest at a low CO2uptake and then reduced as more molecules were anchored on the surface, and interaction between adsorbates and adsorbents was the largest at low surface coverage due to presence of unsaturated active sites. For instance, Yuanet al[11]reported an initialQstof 29.5 kJ/mol for CO2adsorption on a polymer-derived N-doped microporous carbon,which decreased to 22 kJ/mol as CO2surface coverage increased. The presentQsttrend for CO2adsorption had some difference to these previously reported results[32], andQstat low CO2uptake was in the middle range with moderate reduction at first and then reversely increased as the further increase of CO2amounts on the surface. The loweredQsttrend at first was caused by saturation of active adsorptive sites from limited N-species, and the later increasedQstas an increase of molecules was because of the occupation of mesopores and increased repulsive interactions of adsorbate-adsorbate, and thus reversely improved adsorbate-adsorbent interaction[39]. It should be noted that AC-5-700 and AC-5-800 from activation had much largerQstas compared to that of AC-0-700 and AC-0-800 without salt in the hydrothermal procedure, indicating the positive effect of N-species for CO2capture from consolidation effect of salt as large mesopores indexed from PSD as shown in Fig. 6. All the results indicated that the moderate level ofQstvalues were beneficial for CO2capture on the chitosan-derived carbons.
4 Conclusion
Chitosan was used as the precursor for the production of hydrochar under salt assistance and converted into carbons by the further mild activation. Activation using two-folds of KOH to char made carbons with developed porosity, showing surface areas and pore volumes of up to 2 547 m2/g and 1.39 cm3/g. AC-5-800 with largest surface areas showed the uptake of 2.69 mmol/g, while AC-5-600 with a surface area of 1 249 m2/g showed the highest CO2uptake of 4.41 mmo/g at 25 °C and 0.1 MPa, indicating both surface area and total pore were not sole factors in determining the uptakes. The CO2uptake depended not only on surface areas but on the amounts of active sites and PSDs especially for pores from the narrower micropores and N-species. The low-cost carbons with excellent CO2uptake form chitosan indicated that the chitosan hydrochar was a potential precursor for the production of high-performance carbons as the next generation of CO2adsorbents.
Acknowledgements
The authors thank supports from National Nature Science Foundation of China (21506184; 21676227),Natural Science Foundation of Hunan Province(2021JJ30665;2019JJ50597), Key Laboratory of Biobased Materials (BMF2020015), and 2011 Collaborative Innovation Center of Chemical Process with Environmental Benignity & Efficient Resource Utilization of Hunan Province.
Note:
The authors declare no competing financial interest.

- 新型炭材料的其它文章
- Preparation of a porous carbon from Enteromorpha prolifera with excellent electrochemical properties
- A DFT study of the effect of stacking on the quantum capacitance of bilayer graphene materials
- 基于碳化钽涂层改性碳基材料的研究进展
- Preparation of a N-P co-doped waste cotton fabric-based activated carbon for supercapacitor electrodes
- Coating a Na3V2(PO4)3 cathode material with carbon to improve its sodium storage
- Two-dimensional layer materials for highly efficient molecular sensing based on surface-enhanced Raman scattering