
骨肉瘤中circRNA-miRNA-mRNA相关调控网络的构建
2021-12-27 08:39王兆强裴露萍
中国矫形外科杂志 2021年23期
王兆强,裴露萍,李 刚
(山东省滨州市博兴县人民医院,山东博兴县 256500)
环状RNA(circular RNAs,circRNA)是一类长的非编码RNA分子,它们形成共价闭合的环状结构,没有5′-3′极性和polyA尾,不受RNA外切酶影响,表达稳定[1]。近几年研究表明,哺乳动物细胞中存在成千上万种内源性circRNA,它们通过充当miRNA海绵与microRNA(miRNA)相结合,阻断miRNA对下游靶基因的降解作用,从而使靶基因表达水平升高,被称为竞争性内源RNA(competing endogenous RNAs,ceRNA) 机制[2,3]。越来越多的研究证实,体内circRNA表达丰度的改变可以通过ceRNA机制影响下游靶基因的表达,从而导致细胞生理功能失调,在肿瘤的发生、发展和转移中发挥重要作用[2]。
骨肉瘤(osteosarcoma,OS)是最常见的原发性恶性骨肿瘤,多发于四肢,而很少见于脊柱,在青春期和60岁以上的人群中有2个发病高峰[4,5]。尽管多药化疗和手术切除原发性肿瘤等医学技术已经改善,但由于骨肉瘤早期转移的特征,其生存率仍然很低[6]。因此,寻找有效的用于骨肉瘤早期诊断的分子标志物及优化个体化治疗是临床亟待解决的问题。越来越多的研究表明,由非编码RNA介导的关键信号通路的分子变化对骨肉瘤的发生发展机制(包括细胞生长、侵袭和转移)至关重要[7]。因此,本实验通过GEO数据库筛选出骨肉瘤中差异表达的circRNAs,miRNAs和 mRNAs,再构建 circRNA-miRNA-mRNA调控网络,对其进行功能和通路富集分析,最后筛选出与骨肉瘤患者预后相关的mRNAs。有望为骨肉瘤的早期诊断开发新的标志物以及为骨肉瘤的治疗开发新的治疗靶点。
1 材料与方法
1.1 数据收集
通过GEO数据库(http://www.ncbi.nlm.nih.gov/geo/) 下载 GSE140256,GSE28425,GSE36001 和GSE28424数据集中基因表达谱数据。在GSE140256数据集中,对3例骨肉瘤组织和3例正常骨组织进行circRNA数据分析;在GSE28425数据集中,对19种骨肉瘤细胞系和4例正常骨组织进行miRNA数据分析;在GSE36001和GSE28424数据集中,对19种骨肉瘤细胞系和8例正常骨组织进行mRNA数据分析。
1.2 差异表达基因的分析
通过edgeR的R/Bioconductor包,设置|log2FC|的临界值,进行差异表达分析,以筛选骨肉瘤和正常组织之间差异表达的circRNA,mRNA和miRNA。|log2FC|>1(foldchange,变化倍数),P<0.05为差异有统计学意义。
1.3 circRNA-miRNA-mRNA网络建立
通过 StarBase数据库(http://starbase.sysu.edu.cn/)预测circRNA-miRNA间结合情况。通过TargetScan(http://www.targetscan.org/)和StarBase(http://starbase.sysu.edu.cn/)数据库预测miRNA的mRNA靶标。基于circRNA/miRNA和miRNA/mRNA的相互作用,利用Cy⁃toscape(3.5.1版)构建circRNA-miRNA-mRNA网络。
1.4 功能分析
利用 Cytoscape(3.5.1版)中的 BiNGO和ClueGO插件进行功能和通路富集分析,GO(gene ontology) 和 KEGG (kyoto encyclopedia of genes and genomes)富集分析评估网络中过表达mRNAs的潜在生物学功能和相关信号通路(P<0.05)。
1.5 基于GEO数据的生存分析
下载GEO中GSE 39055数据集基因表达谱数据和37例骨肉瘤患者的生存时间及生存状态。ROC(receiver operating characteristic)分析用于确定曲线下的面积。在每个mRNA表达谱的ROC曲线上都可以清楚地观察到(0,1)点,该点可以同时最大化灵敏度和特异性。因此,作者将1 512.3、27 975.15、10 931.25、42.3、552.55表达量作为 NUDT11、FSCN1、MYO10、SDC4、UBE2V2生存分析的最佳截断值。通过Kaplan-Meier生存分析评估这6种mRNAs与患者预后之间的关系。
1.6 统计学方法
使用SPSS统计学软件进行数据统计分析,使用非配对T检验分析骨肉瘤组织(细胞)与正常骨组织之间circRNA的差异表达。使用SPSS软件生存函数中的Cox回归模型进行单因素分析。运用Kaplan-Meier方法进行生存分析,并使用log-rank检验分析mRNA表达与骨肉瘤患者预后之间的相关性。以P<0.05为差异有统计学意义。运用Graphpad Prism 7.0软件作图。
2 结 果
2.1 在GEO数据库中筛选circRNAs
在GSE140256数据集中分析了骨肉瘤和正常骨组织之间circRNA的差异表达。定义变化倍数>2且P<0.05的circRNA为表达差异具有统计学意义,筛选出6个表达上调的circRNAs(hsa_circ_0000006、hsa_circ_0046264、 hsa_circ_0078767、 hsa_circ_0094088、hsa_circ_0096041和 hsa_circ_0049271)。基于上述circRNAs构建表达热图(图1a),circRNA差异表达见图1b~g。
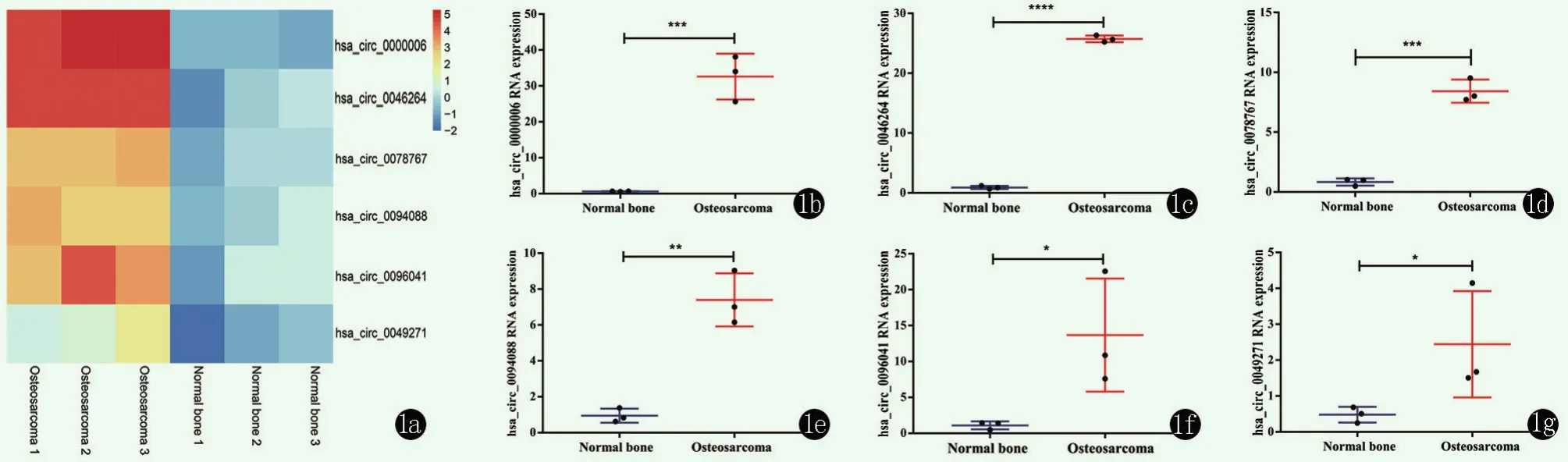
图1 基于GEO数据集(GSE140256)的基因表达分析 1a:在3个OS组织中与3个正常骨组织相比,6个上调的cir⁃cRNAs的热图。每一列代表一个样品,每一行代表一个circRNA。从蓝色(低)到红色(高)的色标指示归一化表达的水平 1b~1g:基于GSE140256数据集的相应circRNAs差异表达图
2.2 在GEO数据库中筛选miRNAs和mRNAs
在GSE28425、GSE36001和GSE28424数据集中分析了骨肉瘤和正常骨组织之间miRNA和mRNA的差异表达。定义变化倍数>2且P<0.05的miRNA和mRNA具有差异表达,筛选出124个表达下调的miRNA;GSE 36001和GSE 28424数据集分别筛选出604和687个表达上调的mRNA,做Venn图筛选出504个共同表达上调的mRNAs(图2a)。

图2 circRNA-miRNA-mRNA网络的富集分析 2a:基于2个GEO数据集(GSE36001,GSE28424),与正常骨组织相比,OS中上调的mRNAs的Venn分析 2b:OS中的circRNA-miRNA-mRNA调控网络。节点颜色代表不同的RNA类型 2c:circRNA-miRNA-mRNA网络中过表达mRNAs富集的前20 GO分析生物学过程。球的大小代表富集的基因个数,颜色代表P值 2d:cir⁃cRNA-miRNA-mRNA网络中过表达mRNAs富集的KEGG通路生物学过程。“*”代表P<0.05;“**”代表P<0.01
2.3 骨肉瘤中circRNA-miRNA-mRNA网络
为了探索circRNA的功能,作者基于ceRNA预测了circRNA-miRNA-mRNA网络内的潜在相互作用。通过StarBase在线工具,预测出能够与hsa_circ_0000006、 hsa_circ_0046264、 hsa_circ_0078 767结合的的下调miRNA分别有4、2、4个(表1)。通过TargetScan和StarBase在线工具,预测出52个能够与上述10个miRNAs结合的上调mRNAs(表1)。根据circRNA,miRNA和mRNA之间的相互作用,使用Cytoscape软件绘制包含3个circRNAs,10个miRNAs和52个mRNAs的ceRNA网络图(图2b)。
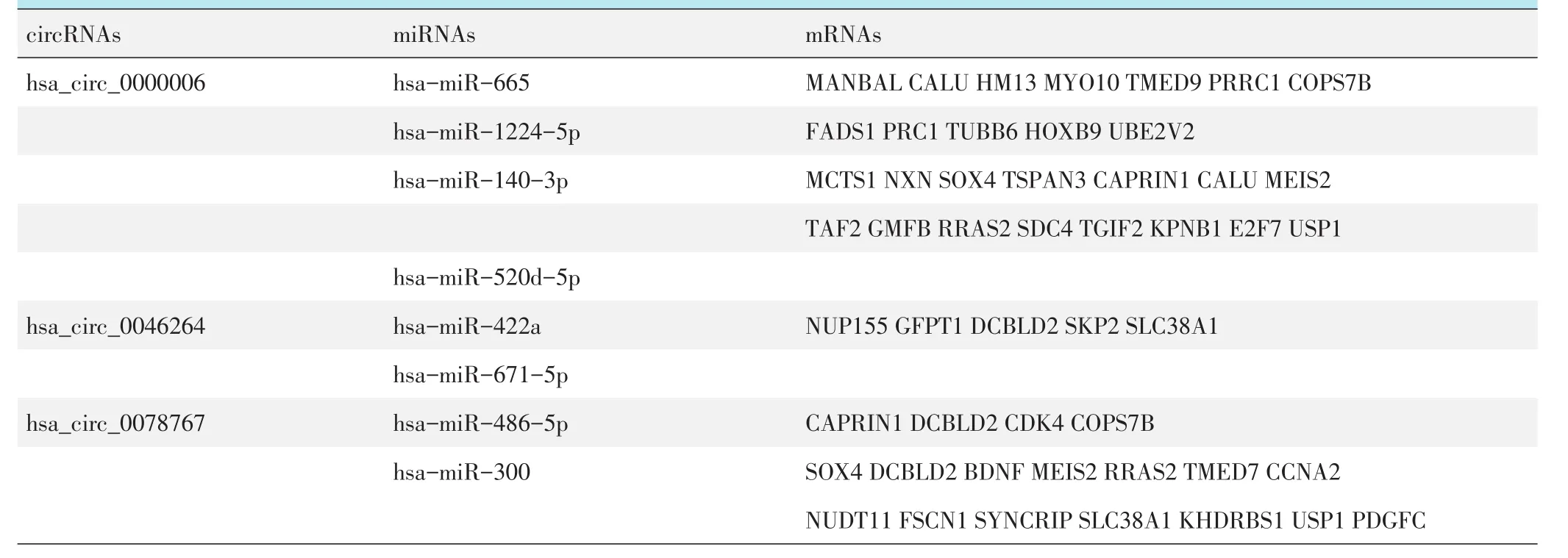
表1 骨肉瘤circRNA相关ceRNA网络中的circRNAs、miRNAs和mRNAs
2.4 circRNA-miRNA-mRNA网络中上调的mRNAs的功能分析
功能分析表明,上述ceRNA网络中52个上调的mRNAs富集20个GO生物学过程类别和8个KEGG类别(P<0.05)。与基因表达失调有关的GO生物学过程是正向调控细胞周期(GO:0048522),正向调控生物学过程(GO:0048518)和正向调控细胞增殖(GO:0008284)(图2c)。根据KEGG分析,p53信号通路和病毒致癌通路与癌症发生有关(图2d)。
2.5 筛选与生存相关的mRNAs
通过Kaplan-Meier生存分析评估了这52种mRNA与骨肉瘤患者预后之间的关系,结果表明,NUDT11、FSCN1、MYO10、SDC4、UBE2V2 过表达患者的总体生存时间显著短于低表达患者(图3)。基于Cox回归模型的单因素分析,发现NUDT11、FSCN1、MYO10、SDC4、UBE2V2过表达均是预后不良的危险因素(表2)。

表2 骨肉瘤中mRNAs的单因素Cox分析

图3 基于GEO数据集(GSE39055)分析circRNA-miRNA-mRNA网络中mRNAs表达与OS患者预后之间的关系 3a~3e:FSCN1、MMYO10、NUDT11、SDC4、UBE2V2过表达患者的总生存时间均显著短于低表达患者(P<0.01,P<0.05,P<0.01,P<0.05,P<0.01)
3 讨论
circRNA是一类新型的非编码RNA,它们可能在基因表达和信号通路中起重要作用,并参与多种疾病的发展。目前研究表明OS中存在特定的circRNA表达失调,其高表达或低表达是OS发生、发展和转移过程中的重要分子事件,但这些circRNAs的作用机制尚不清楚。差异表达的circRNAs可能作为疾病早期诊断的标志物,也可作为新的潜在治疗靶标。
在本研究中,基于GEO数据库,与正常骨组织相比,OS中有 6个 circRNAs(hsa_circ_0000006,hsa_circ_0046264,hsa_circ_0078767,hsa_circ_0094 088,hsa_circ_0096041 和 hsa_circ_0049271)和 504个mRNAs表达上调。据报道,hsa_circ_0000006通过吸附miR-578上调VEGF表达促进骨肉瘤细胞增殖和迁移[8],与本筛选结果一致。有研究发现hsa_circ_0046264通过靶向hsa-miR-1245上调BRCA2的表达抑制肺癌发生发展[9],与本筛选结果不一致,可能与其所在肿瘤微环境有关,有待后续试验进一步验证。其余4个circRNAs尚未有文献报道。
circRNA被证实具有调节mRNA稳定性和蛋白质翻译的能力[2]。作者推测OS中过表达的mRNAs可能受 circRNAs调控。 RPISeq(http://pridb.gdcb.ia⁃state.edu/RPISeq/)和LncTar(http://www.cuilab.cn/lnctar)预测发现没有与6个circRNAs直接结合的mRNAs和蛋白。因此,作者认为circRNAs可能间接作用于靶基因。据报道,miRNA与其靶基因的3'UTR区结合,降低了mRNA的稳定性或抑制蛋白质翻译[10]。ceRNA假说认为circRNA吸附miRNA,从而降低了其丰度并影响了下游靶基因的表达[2]。作者通过GEO数据库筛选了在OS中下调的miRNAs,根据相互间的结合位点确定了潜在的circRNA-miR⁃NA-mRNA相互作用网络,构建了一个完整的ceR⁃NA调控网络。近几年的研究表明,circRNA基于ceRNA机制可能在多种类型的癌症中发挥作用[11~17]。在本研究中,hsa_circ_0000006、hsa_circ_0046264、hsa_circ_0078767结合OS中下调miRNAs分别有4、2、4个。有研究发现,miR-665通过直接靶向HMGB1并激活Wnt/β-catenin途径来抑制视网膜母细胞瘤的致癌性[18];miR-140-3p的高表达可抑制乳腺癌的增殖和迁移[19];miR-671-5p通过阻断细胞周期来抑制骨肉瘤细胞增殖[20];hsa-miR-486-5p通过TGF-β/SMAD2信号通路抑制非小细胞肺癌的迁移和侵袭[21],与本筛选结果一致。作者预测发现,能够与10个miRNAs结合的mRNAs中,NUDT11、FSCN1、MYO10、SDC4、UBE2V2 过表达患者的总体生存时间明显短于低表达患者。基于Cox回归模型的单因素分析,发现NUDT11、FSCN1、MYO10、SDC4、UBE2V2过表达均是预后不良的危险因素。FSCN1促进骨肉瘤细胞的增殖和转移,其高表达与患者预后不良有关[22];SDC4的高表达与远处转移的发生和肿瘤的大小有关,其高表达患者的总体生存期明显缩短[23],上述研究与本分析结果一致。此外,功能分析表明,这些上调的mRNAs可能与肿瘤的发生发展有关,目前对包括OS在内的多种肿瘤均有报道,证实SOX4、FSCN1、SKP2可促进OS 细胞的增殖[22、24、25]。
综上所述,作者确定了3个circRNA-miRNA-mRNA信号轴,circRNA相关的ceRNA网络可能为OS发生发展的分子机制提供有价值的数据支持。由于此分析仅基于生物信息学方法,因此还需要进一步的实验研究,以验证这3条信号轴在OS的发生发展中发挥重要的调控作用。
猜你喜欢
学苑创造·A版(2020年12期)2020-01-07
中国外汇(2019年15期)2019-10-14
山东陶瓷(2019年2期)2019-02-17
实用口腔医学杂志(2017年6期)2017-09-19
中国现代医生(2016年11期)2016-09-06
作文教学研究(2016年1期)2016-07-05
中国病理生理杂志(2015年8期)2015-12-21
吉林大学学报(医学版)(2015年5期)2015-12-16
医学研究杂志(2015年8期)2015-06-22
医学研究杂志(2015年3期)2015-06-10
