
基于微卫星标记对长江下游鲢遗传多样性现状的分析
2021-12-22 05:56罗宇婷方弟安周彦锋徐东坡彭云鑫张桂宁
南方水产科学 2021年6期
罗宇婷,方弟安, ,周彦锋,徐东坡,彭云鑫,彭 飞,张桂宁,刘 凯,尤 洋,
(1. 南京农业大学无锡渔业学院,江苏 无锡 214081; 2. 中国水产科学研究院淡水渔业研究中心,江苏 无锡 214081;3. 上海海洋大学/水产科学国家级实验教学示范中心,上海 201306)
鲢 (Hypophthalmichthys molitrix) 又称白鲢,为“四大家鱼”之一,属滤食性鱼类,广泛分布于我国北部黑龙江水系到南部珠江水系的大江大河与湖泊[1]。长江是鲢天然种群资源的重要产地,但因长江经济带的发展、长期的过度捕捞和环境污染等因素,鲢的种苗产量一直呈下降趋势[2]。近年来为加强对长江渔业资源的保护,实行了以“四大家鱼”为主的人工增殖放流,然而增殖放流在提高渔业资源量的同时也会给在江群体的遗传结构和生态系统带来潜在风险[3]。近年来对鲢遗传多样性的评估主要集中于地理群体的比较和增殖放流资源增量评估,如张敏莹等[4]利用微卫星标记对长江下游4个放流鲢群体 (蠡湖、巢湖、洪泽湖和长江无为段)的遗传分析表明下游群体遗传多样性较丰富,群体间的遗传分化程度较弱;朱晓东等[5]通过微卫星分析长江中下游5个鲢群体 (石首、监利、湘江、九江和安庆) 的遗传多样性结果表明中下游鲢可能存在2个祖先群体;以及对长江中上游鲢群体[6-8]的遗传研究表明上游群体和中游群体分属于长江水系2个不同种群。对增殖放流效果评估的研究,杨习文[3]使用微卫星标记评估2016—2017年长江江苏段鲢增殖放流效果发现,长江江苏段增殖放流对鲢资源量有较好的补充作用,但使该段鲢群体的遗传结构趋向单一化;陈会娟[9]基于D-loop和微卫星分析2015—2017年放流亲本对长江中游家鱼早期资源的影响发现,多年的增殖放流并未对渔获物群体的遗传多样性和遗传结构造成显著影响。以上研究均主要集中于长江中上游、下游零星江段的遗传多样性评估,对放流后长江下游鲢群体遗传多样性的系统评估尚未见报道。
鲢作为典型的江湖洄游性鱼类,每年繁殖季节(4—7月) 在长江干流的产卵场进行繁殖,产后的亲鱼和卵发育成的幼鱼进入鄱阳湖摄食肥育,秋末冬初又回到干流越冬[10]。长江下游作为鲢的重要育幼场所,生境条件优越、生物资源量大、生态价值突出;其中湖口江段连通长江和鄱阳湖,其水域复杂、生境特殊,为鲢的江湖洄游提供关键通道,在长江鲢群体生活史完成过程中发挥着重要的生态功能[11]。因此,本研究基于成熟的微卫星标记技术,使用前人已发表的11 对特异性引物,研究长江湖口及以下共8个江段鲢群体的遗传多样性和遗传结构,评估种质资源状况,了解在经过禁渔期、增殖放流和生态调度等修复措施后在江鲢的种群现状,并与其他湖泊、水系结合比较,更好地为长江下游鲢资源修复和“长江大保护”战略提供科学依据。
1 材料与方法
1.1 样品采集
2017—2019年在长江下游8个江段定制张网,在鱼类调查中随机选取鲢327尾,剪取活鱼的尾鳍5 g左右并保存于无水乙醇中,具体样本分布信息见表1、图1。
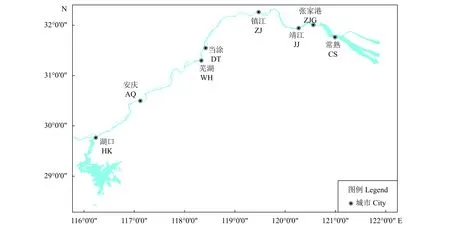
图1 长江下游鲢采样位点Figure 1 Sampling sites of H. molitrix in lower Yangtze River
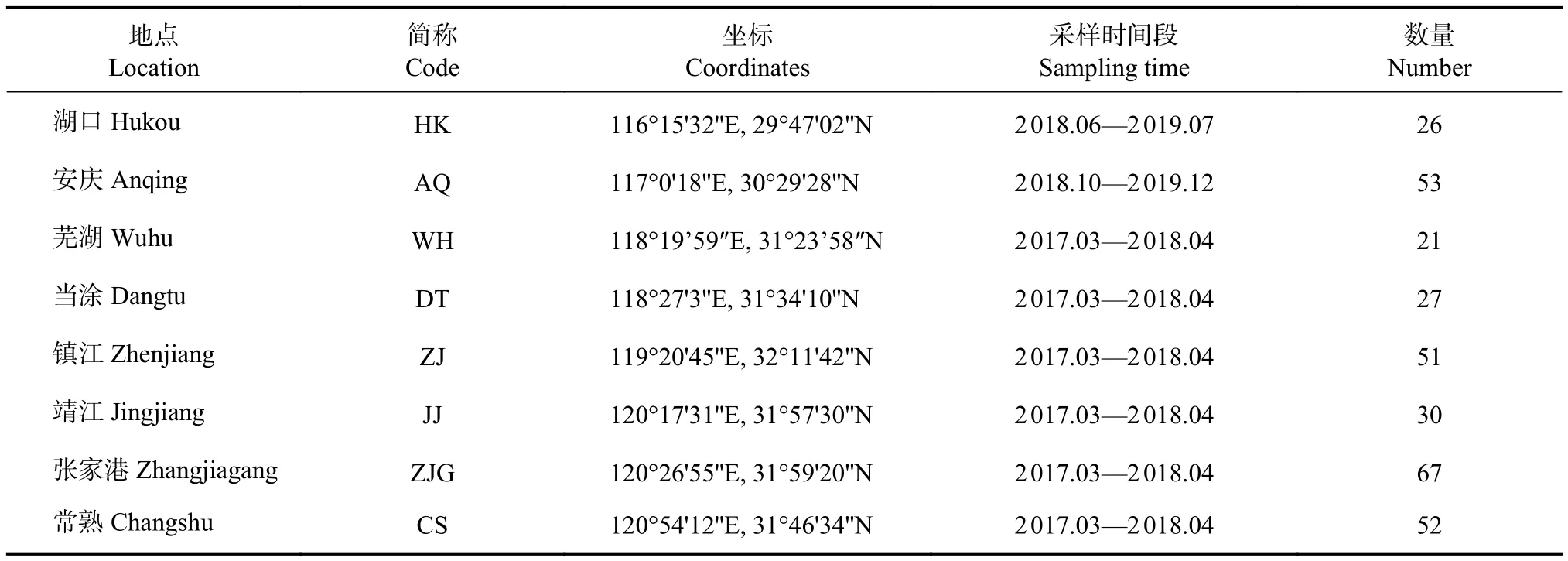
表1 长江下游鲢采样情况Table 1 Sampling of H. molitrix in lower Yangtze River
1.2 DNA提取和PCR
采用试剂盒法提取鳍条组织中的DNA (海洋动物组织基因组DNA提取试剂盒,北京天根生物技术有限公司),1%琼脂糖凝胶电泳检测其质量,于-20 ℃保存。
参照杨习文等[12]方法选取11 对荧光微卫星引物[由生工生物工程(上海)股份有限公司合成]。
10 μL PCR反应体系:Premix Taq (TaKaRa Taq Version 2.0 plus dye) 5 μL,上下游引物稀释液(浓度为 10 μmol·L-1) 各 0.1 μL,DNA 1 μL,去离子水 3.8 μL。
PCR反应程序设定:94 ℃预变性120 s;94 ℃变性 20 s,59~61 ℃ 退火 20 s,72 ℃ 延伸 20 s,反应循环30 次;72 ℃延伸600 s;4 ℃保存。
PCR产物使用2%琼脂糖凝胶电泳检测,通过G:BOX全自动凝胶成像系统观察目的条带成像质量,合格后送至生工生物工程 (上海) 有限公司进行毛细管电泳测序,利用自动测序仪ABI Prism3 730xl (Rox-500 standard) 对扩增产物进行分型。
1.3 遗传多样性参数计算
1.3.1 微卫星位点多态性 基于毛细电泳技术进行基因分型后人工核对基因型准确性,将检测到的微卫星分析信息录入到Excel 2010软件中并转化为文本。利用谱系分析软件Cervus 3.0.7[13]分析11个微卫星位点等位基因数量 (Numbers of allele,Na)、观测杂合度 (Observed heterozygosity,Ho)、期望杂合度 (Expected heterozygosity,He) 和多态信息含量(Polymorphic information content, PIC) 并检测哈迪-温伯格平衡 (Hardy-Weinberg equilibrium,HWE) 偏离情况。
1.3.2 遗传多样性和遗传分化指数 利用GenAlEx 6.503[14]计算各群体的Na、有效等位基因数 (Effective numbers of allele, Ne)、私有等位基因数(Numbers of private alleles, Ar)、He、Ho,无偏期望杂合度 (Unbiased expected heterozygosity, uHe)、Shannon's信息指数 (Shannon's information index, I) 和群体内部近交系数 (Fixation index, F);同时,采用自助法分析 (Bootstrapping analysis,1 000次重复抽样) 评估群体间的遗传分化,通过评估其显著性来计算Fst(F-statitics values, Fst) 的成对估计值和成对的基因流 (Gene flow, Nm) 估计值。
利用软件Arlequin 3.1[15]对样本遗传变异进行AMOVA (Analysis of molecular variance) 分析,得到群体间和群体内的变异水平差异,同时计算成对遗传距离 (Genetic distance, D);应用软件MEGA 5.0[16]基于遗传距离,采用非加权分组平均法 (Unweighted pair-group method with arithmetic mean,UPGMA) 构建系统发育树。
1.3.3 遗传结构 利用软件Structure 2.3.4[17]进行群体遗传结构划分,以贝叶斯模型为基础,将每一个可能的遗传聚类群数目(K)拟设定位1~10,将MCMC (Markov Chain Monie Carfo)开始时的不作数迭代 (Length of burn-in period) 设为50 000次,每个K重复运算5次;采用Evanno等[18]的方法计算Delta K,分析最佳K值为该群体的类群数,通过Clumpp 1.1.2[19]重复抽样分析,由软件GraphPad Prism 8.0.2[20]软件绘制群体遗传结构图。
2 结果
2.1 SSR位点多态性
采用高效毛细管电泳技术将11个微卫星荧光标记的PCR产物进行基因分型 (部分毛细电泳检测结果见图2)。Cervus 3.0.7分析结果显示 (表2),11 个微卫星座位在327个实验样本中共检测出164个等位基因,每个座位的Na介于7 (HLJBL168)~25 (HLJBL217),平均值为14.9;每个座位的Ho介于 0.525 (HLJBL170)~0.850 (HLJBL174),平均值为0.670;每个基因座的He介于0.512 (HLJBL169)~0.936 (HLJBL217),平均值0.773;各位点PIC介于 0.479 (HLJBL169)~0.930 (HLJBL217),均值为0.744,Botstein等[21]研究表明PIC≥0.5为高度多态性,本研究11个位点中除HLJBL169的PIC略低于0.5,其他位点均是微卫星理想的微卫星选择标记,可用于鲢群体遗传多样性的评估。HLJBL165、HLJBL168、HLJBL169、HLJBL176、HLJBL203总体水平未偏离HWE,HLJBL184轻微显著偏离HWE,HLJBL174、HLJBL217中等显著偏离,其他位点均极显著偏离HWE,这表明Na在8个种群中分布不平衡或存在差异,可能与基因的突变、杂交有一定关系[5]。
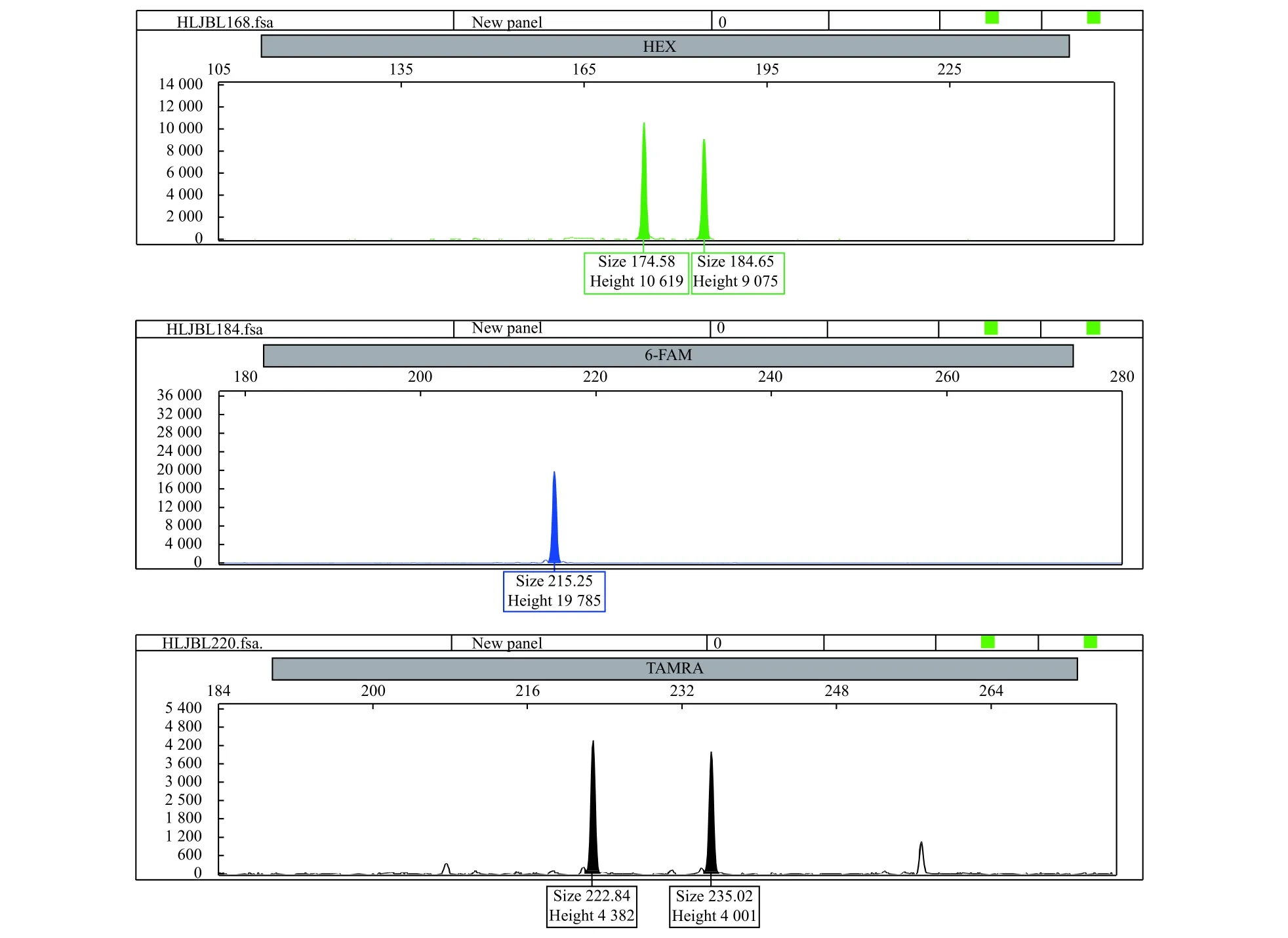
图2 微卫星位点HLJBL168、HLJBL184、HLJBL220基因分型结果Figure 2 Genotype of microsatellite loci HLJBL168, HLJBL184 and HLJBL220
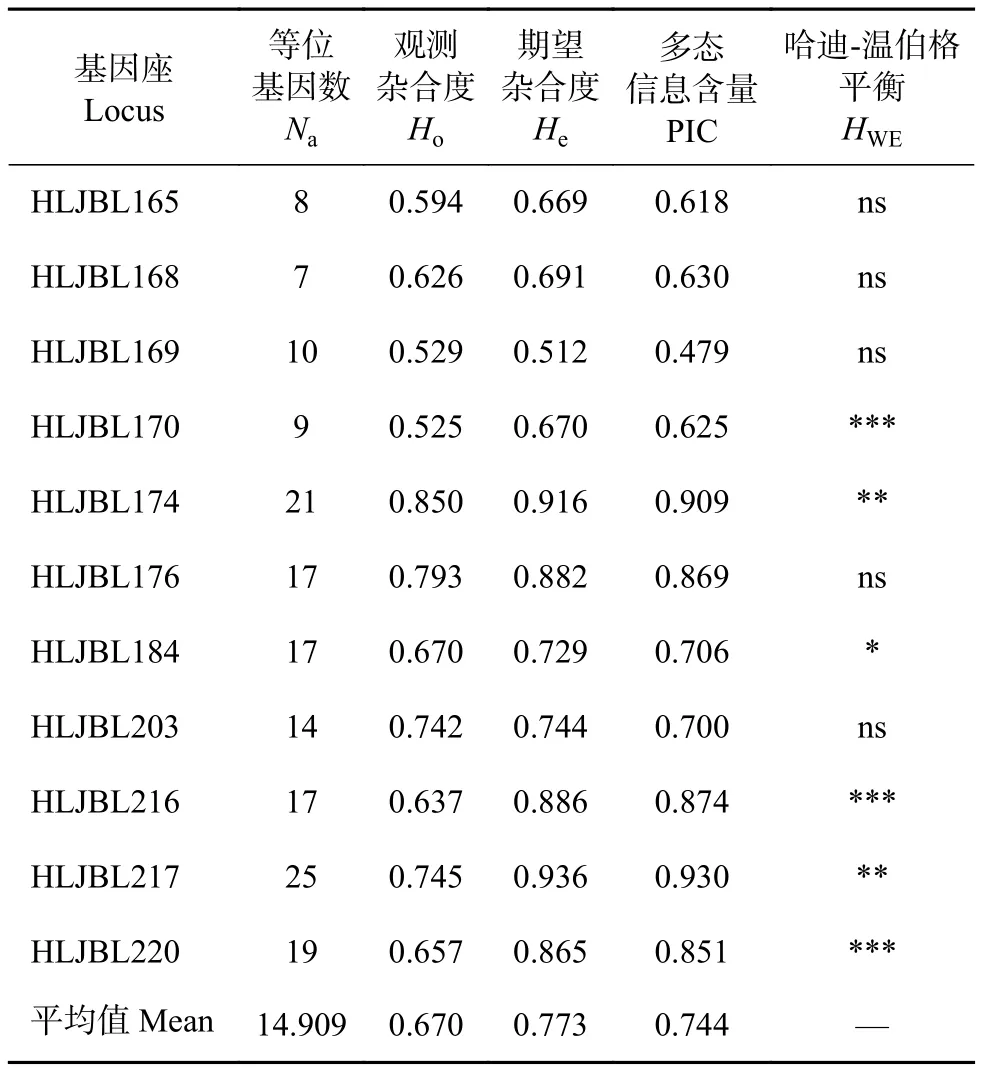
表2 基于11个微卫星标记的鲢遗传多样性水平Table 2 Genetic diversity level of H. molitrix based on 11 simple sequence repeats
2.2 群体遗传多样性分析
遗传多样性结果表明,不同鲢群体的遗传多样性处于不同水平(表3)。各群体Na介于6.00 (HK)~12.3 (AQ),平均值 9.52;Ne介于 3.94 (HK)~6.10 (ZJ),平均值5.38;Ar在AQ群体最高 (17),WH群体最低 (1),均值为5.12;He均值大于Ho的平均值,分别为0.737和0.674,表明纯合子所占比例大于杂合子;所有群体的uHe(即基因多样性指数) 介于0.671 (HK)~0.782 (CS),表明所有种群的遗传多样性均相对较高。I平均值为1.71;群体内近交系数介于0.001 (WH)~0.174 (ZJG),均大于零。
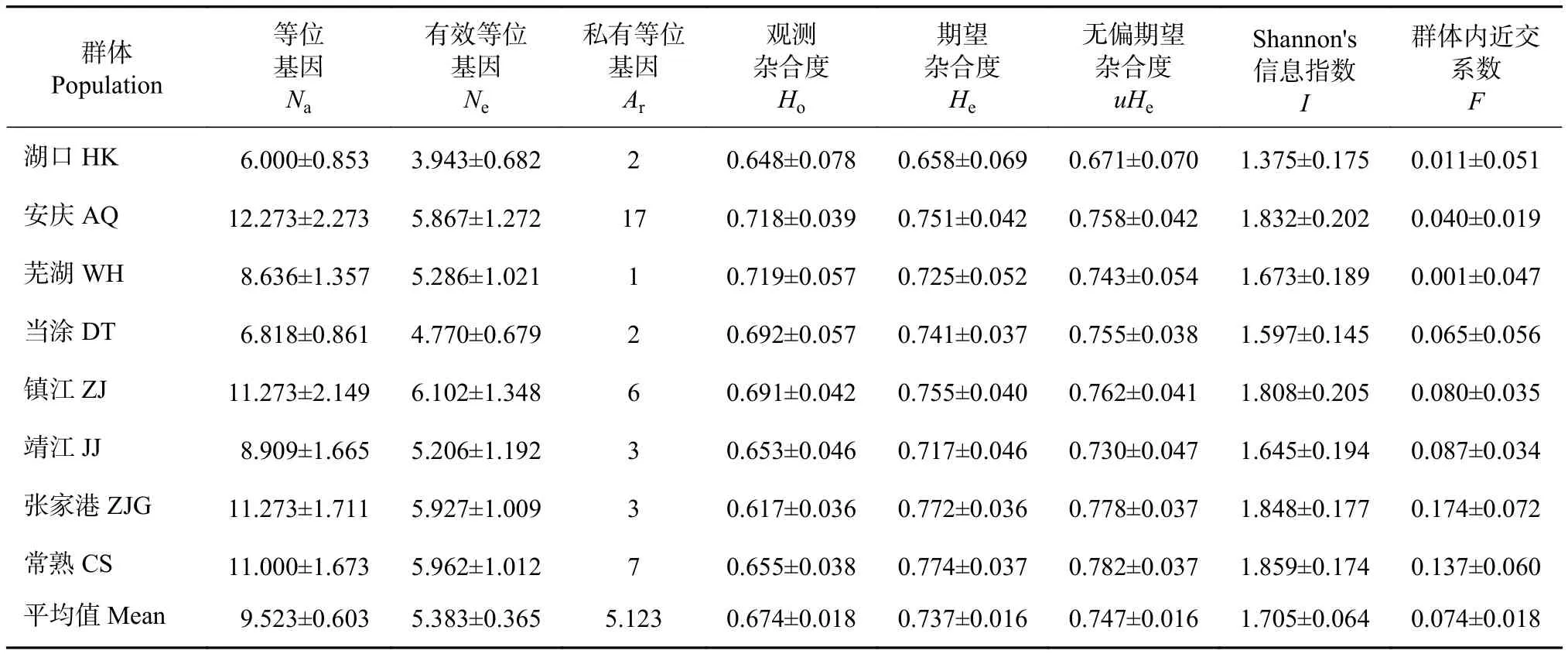
表3 长江下游8个江段鲢的遗传多样性水平Table 3 Genetic diversity of H. molitrix in eight sections of lower Yangtze River
2.3 遗传分化水平
AMOVA分析结果见表4,样本中的遗传变异主要存在于群体内不同个体之间 (97.6%),少部分来源于不同群体间 (2.37%)。计算得出所有群体成对的Fst矩阵和Nm(表5),总体Fst介于0.006(AQ&ZJ)~0.068 (HK&CS),HK群体与其他群体Fst介于0.052~0.068,其他群体间Fst介于0.006~0.021;Nm介于 3.41 (HK&CS)~41.9 (AQ&ZJ),均大于1,表明这8个群体之间能进行基因交流(1<Nm<4),且数值越大基因交流程度越大,除HK外,其他各群体间基因流均大于4,在一定程度上阻止了遗传分化的产生。

表4 基于分子方差分析法 (AMOVA) 的8个鲢群体遗传变异结果Table 4 Molecular variance (AMOVA) results of eight H. molitrix populations

表5 基于微卫星标记的鲢群体Fst (对角线以下) 和基因流 (对角线以上) 配对估计值Table 5 Estimated pairwise Fst (below diagonal) and Nm (above diagonal) of H. molitrix based on simple sequence repeats
各群体间的D介于0.001 (ZJ&JJ)~0.105(HK&CS),ZJ群体与JJ群体D最小 (0.001),HK以下江段 (不包括HK) 的各群体间D均小于0.030;HK群体与CS群体D最大 (0.105),这与两群体地理位置的距离具有一致性。基于D,采用UPGMA (非加权分组平均)法构建了群体系统发育树 (图3),聚类分析结果显示,ZJ和JJ群体首先聚为一类,HK群体与其他群体分为两大类。
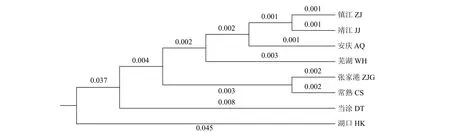
图3 基于遗传距离的长江下游8个鲢群体UPGMA聚类图Figure 3 UPGMA tree based on genetic distance among eight silver carp populations in lower Yangtze River
2.4 遗传结构
绘制DeltaK随K的变化曲线图 (图4),当DeltaK最大时得到最佳K为4,表明所有样本个体最佳可划分为4 个类群 (图5),聚类图中不同颜色代表不同类群,结果显示所有群体的个体遗传结构呈现一定程度的混杂,表明各群体的遗传组成多样化,其中HK群体有明显区分于其他群体的类群1 (黄色图块),该遗传类别在其他群体间也有分布但非常少,表明HK群体有不同于其他群体的基因类群。
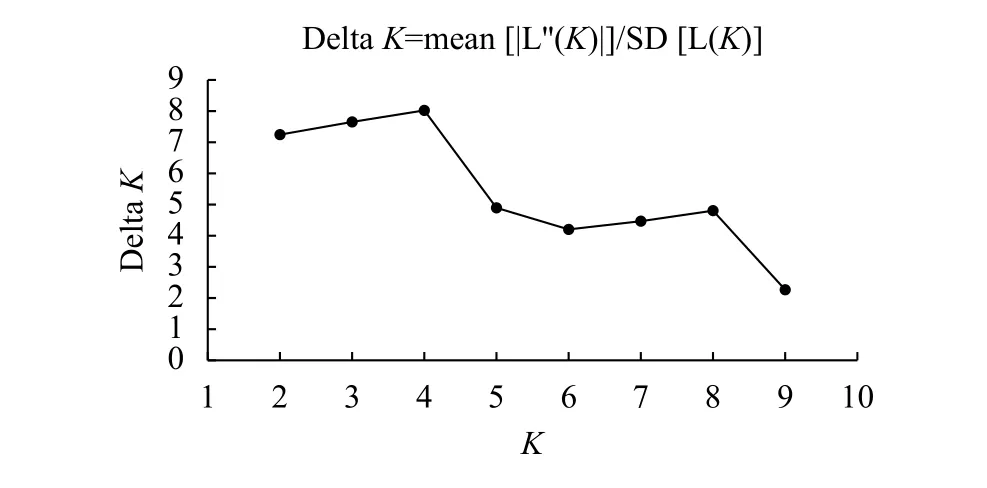
图4 Delta K随K的变化曲线Figure 4 Curve of change of Delta K with changing K value
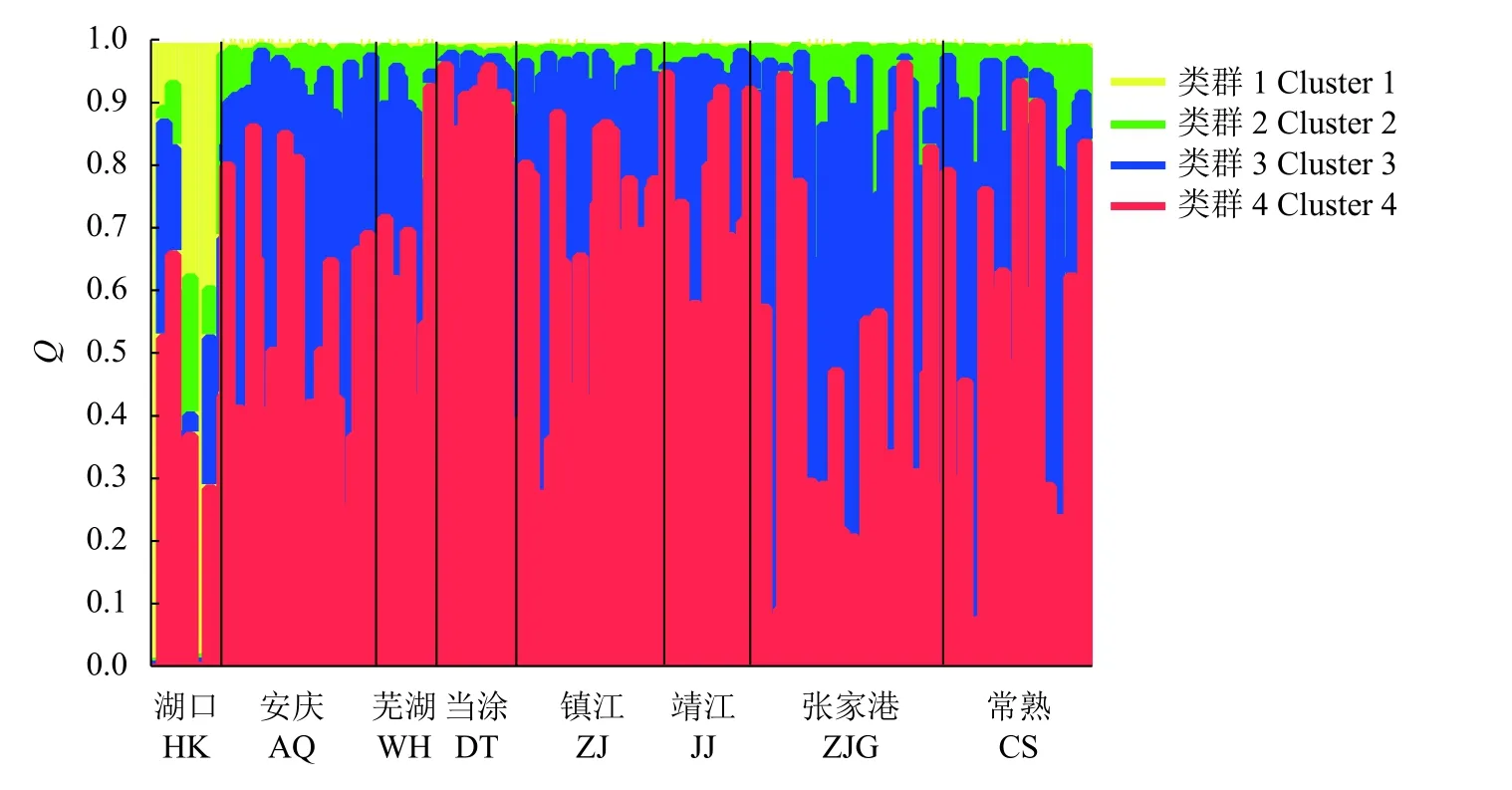
图5 长江下游8个鲢群体聚类结果图 (K=4)Figure 5 Clustering diagram of eight populations of H. molitrix in lower Yangtze River (K=4)
3 讨论
3.1 长江下游江段鲢的遗传多样性
遗传多样性是物种长期生存、适应环境和保持进化的基础,可为种群资源评估提供重要依据[22]。Nm、杂合度I 等参数是反映群体遗传多样性的参考标准,且与基因丰富度和遗传多样性呈正相关[23-24]。本研究利用11对微卫星标记,对长江下游8个江段鲢群体的遗传多样性研究表明,其Na介于6.0~12.3,Ar安庆群体最高 (17),芜湖群体最低(1),但Na多少依赖于样本量大小,因此在样本量差异较大时不宜用Na衡量群体的遗传多样性[25]。生物统计学与抽样调查中通常将30作为区分大样本与小样本的界定值[26]。本研究中由于各采样点捕获量不同,有3个江段 (湖口、芜湖和当涂) 样本数量小于30,但可基本反映该江段鲢群体的遗传资源状况。由于样本量对Na的影响,He更常用于衡量群体的遗传多样性[27]。利用软件进行He的无偏估计,当杂合度介于0.500~0.800即可认为该群体多样性较高[28],结果显示所有群体的uHe介于0.671~0.782。综上,本研究各鲢群体遗传多样性均处于较高水平,且多样性水平表现为:湖口<靖江<芜湖<当涂<安庆<镇江<张家港<常熟,这与I的比较结果 (湖口<当涂<靖江<芜湖<镇江<安庆<张家港<常熟) 接近,其中湖口群体多样性明显低于其他群体,其他群体间多样性水平差异不大。
Na和Ne反映群体遗传变异差异的大小,等位基因在群体中分布的越均匀,Ne就越接近Na[29]。Ho和He的大小反映种群内杂合子的过剩或缺失状态,Ho较大则杂合子过剩,He较大则处于杂合子缺失状态[30]。本研究中所有群体的Na均大于Ne,表明某些等位基因的频率不平均,存在等位基因在群体中分布不均匀的情况;所有群体的He均大于Ho,表明群体内缺乏足够的杂合子,可能存在一定程度的近交现象。Moss等[31]研究表明若F大于0.1,种群会受到近交抑制,根据这一结论推断张家港和常熟两群体内已面临近交抑制的影响。杂合子的缺失和近交现象的存在,可能由长期增殖放流所致[32]。放流鲢作为封闭养殖群体经人工选育,群体遗传多样性一致性较高,种质同质化严重,混入野生群体后基因混杂,使遗传多样性发生改变。
3.2 长江下游江段鲢的遗传分化水平
鱼类在属、种和种群三级水平上的D分别为0.90、0.30、0.05[33],对于长江水系鲢是否属于同一种群,赵金良和李思发[34]用同工酶技术研究1993年长江中下游4个江段 (中游天鹅洲故道、汉阳、瑞昌和下游安庆) 鲢的种群分化表明,4个群体间遗传距离均小于0.001,无显著遗传分化,说明长江中下游水系鲢早期可能有共同的祖先;而李思发和吕国庆[35]用线粒体DNA对1998年长江中下游3个江段 (中游石首、九江,下游芜湖) 鲢的遗传分析发现中下游属于不同类群。两者不同的结论可能是由于中下游代表性采样点或分子标记方法不同。对中游群体[36]、下游4个放流群体[4]的遗传结果均显示出低度遗传分化。而朱晓东等[5]对长江中下游鲢群体的遗传多样性研究表明中游 (石首、监利、九江、湘江) 和下游 (安庆) 之间D介于0.089 3~0.166 5,呈现中偏低的遗传分化。本研究结果显示,湖口群体与下游其他群体间的D介于0.078~0.105,均大于0.05,属于不同种群;而其他7个江段鲢群体间D介于0.001~0.025,小于0.05,隶属同一种群 (表6)。其原因可能是湖口群体与下游群体源于不同祖先,以及地理阻隔、产卵场分布差异造成基因交流贫乏。

表6 长江下游8个鲢群体遗传距离的配对矩阵Table 6 Genetic distance Pairwise matrix of eight H. molitrix populations in lower Yangtze River
Fst是衡量群体间遗传变异程度的可靠参数,Fst<0.05 种群间呈低度分化,0.05<Fst<0.15 种群间呈中度分化[37]。本研究数据显示,湖口群体与其他群体Fst介于0.052~0.068,呈现中等程度的遗传差异;其他群体间Fst介于0.006~0.021,遗传差异很小。除湖口群体外,其他群体间基因流远大于4,说明群体间可随机交配,由遗传漂变引起的群体间变异可能性很小[22],变异主要来自于群体内,这与AMOVA分析结果一致;此外,低遗传分化和强大的基因流,表明它们的地理距离可能非常短或通过某些途径发生基因交换,而各种群间最短距离至少超过50 km,因此推测增殖放流增加了下游群体间的基因交流;然而过度的基因流可能导致瓶颈效应[38],因此有必要控制放流群体的释放规模与苗种质量,维持合适的基因流。
3.3 长江下游江段鲢的遗传结构
本研究UPGMA聚类结果显示湖口群体与其他群体分属于2个种群,8个鲢群体的遗传结构图也显示湖口群体有明显区分于其他群体的基因类群。同一种群的遗传特征是具有一定的基因组成,即共享基因库[34],湖口作为长江中下游分界点,可能分别具有中游和下游的基因库,因而该群体较下游其他群体显示出更大的遗传距离和更多元的遗传结构组成,另一方面也反映了长江中游和下游之间可能存在一定的遗传分化。同时,湖口连通江湖,鄱阳湖作为诸多鱼类的肥育场所,湖泊内生态条件优厚,饵料资源丰富,部分鲢群体可能在湖内完成索饵和育幼阶段的生活史,形成了与下游洄游入湖的群体不同的种群组成。然而长江下游群体间遗传分化很小,这与张敏莹[4]的研究结果相似。究其原因,首先排除地理分布距离远近导致的遗传差异,因下游各江段群体间的遗传差异未呈现出与地理位置的相关性,这与姬长虹等[26]的研究结论一致。研究表明,增殖放流活动或养殖鱼的逃逸可引起野生群体与养殖群体基因参渗,缩小不同地理种群间的遗传差异,增加群体间的基因交流,从而导致下游群体间遗传结构差异较小。综上,湖口江段位于鄱阳湖出水口与长江的交汇处,鱼类活动范围更广泛,结构组成多元化,因此,湖口群体具有与下游其他群体不同的遗传结构组成,造成这种差异的原因是距离上的,还是生境差异的隔离,仍有待进一步探究。
3.4 长江下游江段鲢种质资源状况与保护策略
从等位基因数、杂合度和Shannon's信息指数看,长江下游8个江段整体遗传多样性较丰富,略高于2009年长江(湘江、监利和枞阳段)、珠江、黑龙江野生鲢群体的遗传多样性[26],高于2007年中下游5个群体 (石首、监利、九江、湘江和安庆)[5]和2010年长江下游4个放流群体 (蠡湖、巢湖、洪泽湖和长江无为段)[4],而低于2008年长江中上游2个群体 (万州、监利)[6]和2012年长江 (彭泽段)、赣江、鄱阳湖[39]鲢群体的遗传多样性。可以初步得出长江下游野生鲢的遗传丰富度较上游群体呈衰退趋势,但随着人们对遗传信息重要性的认识和增殖放流评估技术的发展,禁渔禁捕策略的实施和一系列生态调度补充策略,下游鲢种群的遗传丰富度已得到一定程度的恢复。
遗传多样性不仅包括变异水平的高低,也包括变异的分布格局,即种群的遗传结构,是保护生物多样性的重要理论和实际依据。本研究中湖口是连通鄱阳湖与长江的唯一江段,是诸多鱼类完成生殖洄游、索饵洄游等一系列生活史的必经之路,因此湖口江段鲢群体可能同时具有中、下游和湖泊的鲢基因库,保护该江段的生境与渔业资源才能保证江湖连通的天然性,使鱼类种群顺利完成生殖、肥育。此外,还应掌握长江多个江段鲢的基因库组成,并逐步完善其遗传信息,将科学指导应用到实际生产中,以不同江段鲢的遗传信息和遗传结构作为参照,使不同流域增殖放流的亲鱼来源于对应流域的天然种群,避免基因混杂带来潜在的遗传风险,建立人工增殖放流长效机制,从根源保护物种的多样性。
4 结语
本研究基于11个多态性微卫星位点,分析了长江下游多个江段鲢的遗传多样性现状,系统了解下游鲢的整体遗传水平,表明江湖连通对于鲢资源的修复和繁衍具有重要的生态学意义。后续将进一步开发鲢的微卫星位点,或结合其他分子标记方式,摸清各年度长江水系鲢的遗传多样性与变异情况,同时结合放流亲本分析,掌握种群遗传动态,从而科学制定原良种场亲鱼收集原则和增殖放流策略,为长江鲢的物种保护和长江生态系统维稳提供科学依据。
猜你喜欢
今日农业(2022年15期)2022-09-20
烟台大学学报(自然科学与工程版)(2022年3期)2022-06-30
水产科学(2022年2期)2022-03-20
南京师范大学学报(工程技术版)(2021年2期)2021-10-20
船海工程(2020年2期)2020-06-08
科学导报(2020年27期)2020-05-13
四川动物(2020年1期)2020-02-27
生物学教学(2018年3期)2018-08-08
中学生物学(2018年8期)2018-03-01
中学生物学(2008年6期)2008-08-29
