
活化分子氧降解水中典型新污染物研究进展
2021-12-21 04:39杨林峰刘佳佳及吉祥沈铸睿
环境科学研究 2021年12期
李 惠, 杨林峰, 刘佳佳, 及吉祥, 沈铸睿
南开大学材料科学与工程学院, 天津 300350
近年来,新污染物(ECs)在水体中被频繁检出,严重威胁着生态环境安全和人类健康. 如可以引起人体生殖性疾病,包括生育力降低、流产率更高、乳腺癌、前列腺癌和睾丸癌,内分泌失调,可能导致某些生态系统的灭绝等[1-2]. 目前,ECs的处理技术主要包括活性炭吸附法、膜过滤法、高级氧化技术(AOPs). 其中,相较于活性炭吸附法和膜过滤法,AOPs具有反应速度快、降解能力强、无二次污染等优点. AOPs逐渐发展为一种有前景的环境修复方法. AOPs是指在高温、高压、电、光、声等条件下将外部氧化剂[3-5](O3、H2O2、过硫酸盐等)转化为活性氧物种(ROS),如羟基自由基(·OH)等,进而将有机污染物进行矿化降解. 其中,相较于其他氧化剂,分子氧(O2)在空气中的含量高达21%,是绿色、储量丰富且廉价的氧化剂,可被催化转化为(ROS),对有机污染物进行氧化降解,在此过程中生成的ROS主要包含·OH、超氧阴离子自由基(·O2-)、过氧化氢(H2O2)及单线态氧(1O2). 但由于O2自旋禁阻效应,在温和条件下,有机污染物不能被O2直接氧化. O2可通过以下两种路径进行活化: ① O2可被光电子或热电子还原生成ROS; ②三线态O2可获取外部能量转化为1O2[6]. 另外,活化O2主要依赖水中的溶氧,O2经由扩散,吸附在催化活性中心上,才能得失电子生成ROS. 由于活性中心位置和数量的限制,其ROS生成效率普遍不高. 且活化O2过程中电子利用效率普遍不高,不利于新污染物的高效降解. 因此,开发新型高效光催化剂用于活化O2降解新污染物成为必然趋势. 活化O2用于降解新污染物将为环境治理提供一定的借鉴意义.
1 分子氧活化机理
O2大量存在于空气中,是最绿色、环保、经济的氧化剂. 在污染物的去除降解过程中,绝大多数都需要氧化剂的参与. 然而,常态下,O2的HOMO轨道上有两个自旋未成对的电子,为自旋三重态. 而有机物分子的HOMO轨道上一般充满自旋配对的电子,为自旋单重态. 根据电子跃迁选择定则,单重态到单重态和三重态到三重态跃迁为自旋允许跃迁,单重态到三重态为自旋禁阻跃迁. 因此,O2与绝大多数有机分子之间不能直接发生反应. 鉴于此,需要将O2活化,从三重态激发到高能态,来催化降解有机污染物.
O2活化过程通常通过得到电子弱化氧氧键,转化为ROS. O2活化过程中主要产生4种主要的ROS:·O2-、H2O2、·OH和lO2. O2单电子还原过程如图1所示,O2得到电子可被逐步还原为·O2-、H2O2、·OH、H2O.
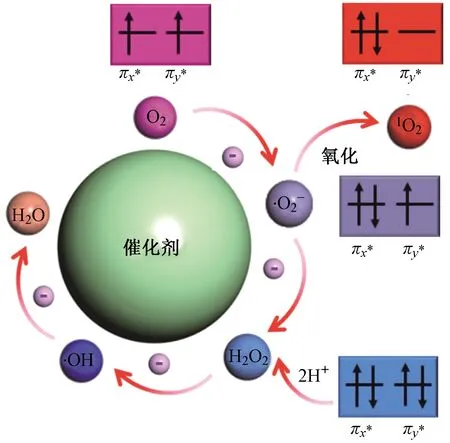
图1 O2活化机理示意[7]Fig.1 Molecular oxygen activation mechanism diagram[7]
首先,O2获得一个电子填充一个π*轨道活化为·O2-,之后得到一个电子填充另一个π*轨道,再通过质子化π*轨道生成H2O2,进一步得到一个电子通过O—O键的断裂生成OH,lO2可由·O2-氧化得到. 反应方程式及电极电势(pH=7)如下[8]:
O2+e-→·O2-, ε=-0.33 V
(1)
·O2-+2H++e-→H2O2, ε=+0.89 V
(2)
H2O2+H++e-→H2O+·OH, ε=+0.38 V
(3)
·OH+H++e-→H2O, ε=+2.31 V
(4)
2 主要的活性氧物种
2.1 超氧阴离子自由基(·O2-)
·O2-中氧的氧化数为-1/2, 是一种中度反应强度、半衰期约为3.1~3.9 μs的ROS[9]. ·O2-是O2单电子还原的产物〔见式(1)〕,其中,电子可来源于零价金属(如Cu、Fe、Al等)、低价金属离子(如Fe2+、Cu+等)、酶存在下的有机底物、原子团簇、光生电子等. 在环境领域,它可被考虑用作氧化剂的作用,同时也不排除作为还原剂的可能性. 尽管其氧化性不是很强,但是·O2-与很多其他活性氧产生途径密切相关,在高级氧化过程中起着重要作用.
2.2 过氧化氢(H2O2)
H2O2是一种重要的绿色氧化剂,广泛应用于各种行业,是一种有前景的清洁燃料,应用于喷气式汽车和火箭. 目前,H2O2的合成主要采用高能耗的间接蒽醌法. 催化活化O2合成H2O2已成为一种安全、环保节能的工艺. 在所有的ROS中,H2O2是唯一相对稳定的分子. 它的氧化电位为1.763 V(pH=0)和0.878 V(pH=14). 在O2活化过程中,O2通过连续单电子还原〔见式(1)(2)〕或双电子还原过程〔见式(5)(6)〕生成H2O2,电子来源为零价金属、低价金属离子、光生电子、电流等.
O2+2e-→O22-
(5)
O22-+2H+→H2O2
(6)
2.3 羟基自由基(·OH)
·OH具有极高的氧化能力(2.8 V),半衰期约10-9s,它可以对大多数有机污染物进行非选择性氧化[10-12]. O2可被零价金属、电流等还原为H2O2,再经过单电子还原过程,还原为·OH. 如Xiao等[13]合成了FeCo合金包裹碳气凝胶材料(FeCoC),最佳金属掺杂比的FeCo合金有利于富氧官能团的原位形成,可通过双电子过程将O2还原为H2O2〔见式(7)〕,碳壳层经单电子过程将H2O2还原为·OH〔见式(8)〕.
O2+2H++2e-→H2O2
(7)
H2O2+e-→·OH+OH-
(8)
另外,原位生产·OH也有其他方法,包括氢取代有机化合物、铁催化、臭氧处理、光催化、电化学、微生物燃料电池、超声分解等[14].
2.4 单线态氧(单重态氧)(lO2)
O2在常态下处于三重态. 而在三重态基态之上有两个激发态lΔg和l∑g+,分子轨道理论也预测了这两种状态. 由于l∑g+状态能量高、稳定性低,寿命比lΔg状态短得多,因此,1O2主要指的是lΔg(lO2)状态. 根据溶剂的不同,1O2在溶液中的寿命约为4~16 000 μs[8],比·OH更长. 直接用电子激发O2到单重态是自旋禁止的,因此,1O2的生成通常用光敏方法或化学方法[15]. 超氧阴离子可被自由基〔见式(9)(10)〕、光生空穴〔见式(11)〕等氧化生成1O2,且概率为2/5[16].
·O2-+·HO2+H+→1O2+H2O2
(9)
·O2-+·OH→lO2+OH-
(10)
·O2-+h+→lO2
(11)
3 分子氧活化策略
ROS可由零价金属、低价金属离子、酶存在下的有机底物、原子团簇、光生电子等提供电子对O2进行活化得到. O2活化过程中的催化材料主要包括酶、零价金属及离子、缺陷半导体、原子团簇及单原子等. 该文将从酶、零价金属及离子、缺陷半导体、负载型原子团簇及单原子催化剂等4种催化材料对以上4种ROS的具体产生途径进行讨论.
3.1 酶催化
在生命体系内部,氧化酶和加氧酶可以在温和条件下活化O2,生成过氧、超氧等ROS参与各种生化反应. 同时也可以活化底物进行加氧反应. 由于酶的作用环境十分苛刻,故科研人员常用小分子金属模型配合物模拟加氧酶活化O2的过程. 含铁和铜活性位点的酶在生物系统中双氧的结合和活化过程中起关键作用. O2只用作氧化剂而未进入产物中的过程是氧化酶催化. 氧化酶生物催化反应与燃料电池类似,为两个半反应,如图2所示,其中SubH2是有机底物,Subox是氧化的有机底物[16]. 加氧酶可以催化底物,将O高效地插入到产物中,同时O2也作为氧化剂[17]. 根据插入底物的氧原子的数目,可以分为单加氧酶和双加氧酶. 典型的单加氧酶有细胞色素P450、甲烷单加氧酶等. 典型的双加氧酶有栋精2,3-双加氧酶等.

图2 氧化酶生物催化反应[16]Fig.2 Oxidase biocatalytic reaction[16]
当双加氧酶的活性中心有还原性金属离子时, 如extrodial型儿茶酚双加氧酶中包含金属辅因子FeⅡ,如图3所示[18-21],FeⅡ可与三重态3O2发生单电子氧化-还原反应生成·O2-和FeⅢ,·O2-与FeⅢ配位生成a中间体S—FeⅢ—·O2-(step 1),之后FeⅢ与配位底物发生快速分子内单电子转移生成FeⅡ,同时底物被活化为S·生成b中间体S·—FeⅡ—·O2-(step 2),此类双加氧酶反应过程金属离子先活化O2再活化底物;当双加氧酶的活性中心有氧化性金属离子时,如introdial型儿茶酚双加氧酶中包含金属辅因子FeⅢ,如图4[22-24]所示,配位底物与FeⅢ在分子内发生快速单电子转移,底物被活化为S·,生成a中间体S·—FeⅡ(step 1),之后O2与S·反应生成b中间体底物—·O2-. 此类双加氧酶反应过程金属离子先活化底物为自由基,然后底物自由基进一步活化O2;对于一些特定的酶,去质子的有机底物与O2直接发生电子转移,底物与O2同时被活化生成底物自由基和·O2-[25]. Decker等[26]研究了血红素、非血红素铁和铜酶激活双氧催化脂肪族的反应,提出了不同的双氧激活方法和机理. Laskavy等[27]发现了一种钌双联吡啶化合物通过双加氧酶途径可以用于O2的活化,其形成的[Ru](O)2中间体是氧原子的供体.

图3 Extrodial型儿茶酚双加氧酶中氧气与底物的活化机理[18-21]Fig.3 Dioxygen and substrate activation mechanism proposed in extrodiol catechol dioxygenase[18-21]

图4 Introdial型儿茶酚双加氧酶中氧气与底物的活化机理[22-24]Fig.4 Substrate and dioxygen activation mechanism proposed in extrodiol catechol dioxygenase[22-24]
由于酶大多数存在于生命体的内部,与生物体有关. 因此,相较于环境科学领域,酶催化O2活化更多地在生物化学领域中被研究.
3.2 零价金属
3.2.1零价铁
零价铁(ZVI)具有很强的还原性,可被用于修复地下水和土壤污染. 然而,利用其还原性直接还原污染物的方法因有很多缺点而饱受争议[28]. 鉴于此,ZVI活化O2降解污染物的方法受到青睐. ZVI与空气中的O2反应后,被氧化为对应的高价铁,而O2被还原为ROS,进而进行后续反应. O2还原过程有两种途径:一种是O2直接得到两个电子生成H2O2〔见式(12)〕[29-30];另一种是O2与二价铁反应,生成·O2-,其继续与二价铁反应,生成H2O2〔见式(13)(14)〕[31]. 紧接着,生成的H2O2会与二价铁发生Fenton反应生成·OH〔见式(15)〕.
(12)
FeⅡ+O2→FeⅢ+·O2-
(13)
·O2-+2H++FeⅡ→H2O2+FeⅢ
(14)
FeⅡ+H2O2→FeⅢ+·OH+OH-
(15)
虽然铁/氧气体系可以活化O2降解污染物,然而,此反应的电子利用率极低. 有研究[32]表明,纳米零价铁活化O2生成·OH的效率仅为理论值的10%左右. 这可能是因为体系反应复杂,有较多副反应导致的. 如ZVI的钝化、H2O2的还原、H2的产生等,从而导致ROS的产量大大减少[33-34]. 因此,很多学者对ZVI活化O2体系进行了改进,如构建双金属[35-38]、核壳结构[39-41]、加入配体[42]等.
3.2.2零价铜
除了铁物种之外,铜物种在O2的活化过程中也显示出优异的活性. 虽然Cu的还原能力比Fe弱,但是Cu+/Cu(E0=0.522 V),Cu2+/Cu(E0=0.341 V)和Cu2+/Cu+(E0=0.16 V)电对的标准电极电势均负于O2/H2O2(E0=0.695 V),表明零价铜(ZVC)在热力学上可以将O2还原为H2O2.
Cu和Cu+均可以将O2有效转化为ROS. 越来越多的研究表明,Cu+在O2的活化过程中起着主要作用. Cu+不仅可以将O2转化为·O2-和H2O2,还可以与H2O2构成类Fenton体系,生成高活性的·OH〔见式(19)(22)(23)〕[43-46]. 在Cu活化氧气的系统中,Cu被认为主要起到生成Cu+和通过双电子还原O2生成H2O2的作用〔见式(16)〕[47-48],其也会生成一定的Cu2+〔见式(17)〕. 而Cu通过单电子还原将O2还原为·O2-的过程最近被证实存在. 有学者报道了在酸性条件下降解双酚A的过程中,Cu与O2直接反应生成·O2-,并且还催化H2O2生成·OH[49]. Zhang等[48]研究发现生成的·O2-促进了Cu2+还原为Cu+〔见式(21)〕. 倪永炯等[50]在降解恩诺沙星时发现,溶液酸性越强,Cu/O2的氧化能力越强. 另外,由于Cu2+/Cu+的E0比Cu+/Cu和Cu2+/Cu的E0更负,因此Cu+转化为Cu2+在热力学上是可行的〔见式(19)〕. 由于铜离子比铁离子适应的pH范围更宽,因此有利于类似Fenton反应中Cu(Ⅰ)与Cu(Ⅱ)的循环〔见式(18)(20)〕.
2Cu+O2+2H2O→2Cu++H2O2+2OH-
(16)
Cu+O2+2H2O→Cu2++H2O2+2OH-
(17)
Cu++H++H2O2→Cu2++·OH+H2O
(18)
2Cu++O2+2H+→2Cu2++H2O2
(19)
Cu2++H2O2→Cu++2H++·O2-
(20)
Cu2++·O2-→Cu++O2
(21)
Cu++O2→Cu2++·O2-
(22)
Cu++2H++·O2-→Cu2++H2O2
(23)
3.2.3其他金属
除了铁、铜外,其他金属也可以活化O2,如零价铝(ZVAl). 从理论上讲,由于Al3+/Al0对的氧化还原电势比Fe2+/Fe0更负,ZVAl有更高的活化氧能力. 有研究表明,只有在酸性(pH<4)条件下,ZVAl才能还原O2原位生成H2O2,并且产生·OH降解污染物[51]. 由于Al只有一个变价,因此其反应与铁/铜物种相比简单了许多.
关于ZVAl活化O2的降解研究已经有很多,包括4-CP[52]、双酚A[53]、对乙酰氨基酚(ACTM)[54]等. 与ZVI相比(pH<3),ZVAl对pH的适用范围更宽(pH<4). 但氧化铝不易去除,另外,仍需要提高实际氧活化能力来满足应用需求.
除以上金属外,零价锌、零价镍活化O2生成H2O2也有少量报道. 值得注意的是,在实际应用中,应考虑到对应离子的毒性并进行相应的后处理.
3.3 缺陷半导体材料
除了金属之外,半导体在O2活化过程中的作用逐渐显露. 研究表明,O2分子能够吸附在半导体表面,得到光生电子后,转化成ROS. 而表面缺陷的存在是O2吸附的前提条件,常用于O2活化的半导体材料包括金属氧化物、BiOX等.
3.3.1金属氧化物
二氧化钛在1972年被发现有光催化性能[55]. 对于二氧化钛,氧空位是常见的表面缺陷,它可以吸附O2并且将其活化. Li等[56]通过DFT计算发现,氧空位和Ti3+间隙掺杂提供的过量电子有利于O2在TiO2表面的吸附,并且在(101)表面形成·O2-. 除了氧空位缺陷,其他表面缺陷结构如线、台阶、角或扭折缺陷也可以活化O2.
已被证实,O2活化的状态与其在TiO2表面的覆盖度有关:当吸附的O2与氧空位的比例小于或等于1时,O2发生双电子还原,以过氧根(O22-)的形式存在;而当吸附的O2与氧空位的比例大于1.5时,O2发生单电子还原,以·O2-的形式[57-58].
另外,Mao等[59]研究发现有氧空位缺陷的BiO2-x可将吸附在氧空位的O2快速转化为·O2-,且氧空位附近的O取代可起协同作用,产生的ROS对甲基橙有较好的降解效果.
3.3.2BiOX
BiOX(X为F、Cl、Br、I)是一种新型半导体催化剂,它特有的层状结构特征有助于光生电子和空穴的分离,从而提高载流子的利用率. BiOX随着卤素原子序数的增大,带隙递减. 考虑到毒性和带隙宽度,BiOCl和BiOBr更受人们关注.
有研究[60]表明,相对于TiO2,BiOCl纳米片的(001)晶面可以在太阳光下更加可持续地活化O2以去除有机污染物,并提出(001)晶面上的氧空位可以通过紫外线有效地刷新. Zhao等[61]提出,BiOCl的(001)晶面倾向于将O2单电子还原为·O2-,而(010)晶面倾向于将O2双电子还原形成O22-,这是由在紫外光照射下(001)和(010)晶面的表面原子暴露和原位生成的氧空位特性共同作用的. 对于BiOBr,有学者证明了结合的氧空位可以显著加速激子离解,从而增加了电荷载流子的产生,有利于在光催化反应中表现出出色的性能[62].
3.3.3金属硫化物
二硫化钼是一个良好的压电半导体. 在压电催化降解过程中,它能与吸附的O2强烈相互作用,高效地产生ROS. 最近,有学者报道了高硫空位浓度的富缺陷MoS2,用以铬黑T染料的降解. 通过硫空位缺陷诱导催化活性Mo原子,通过引入Mo4+/Mo6+氧化还原对促进了O2的双电子还原,对ROS的生成显著增强[63]. 另外,Sun等[64]发现,有S空位缺陷的β-In2S3可将O2活化生成·O2-,其可将醇高效且高选择性地转化为醛.
3.4 原子团簇/单原子
最近,有学者发现原子团簇/单原子对O2活化也有一定的作用.
有学者研究了中性金团簇(Aun, 4≤n≤21)与O2的相互作用. 其中,电子由金团簇转移到O2中,形成超氧物种. 奇数个数的AunO2(n=7、9、11、21)中形成了电荷转移复合物,从而形成了超氧(·O2-)物种. 相反,偶数个数的团簇除Au4、Au10和Au12外在很大程度上没有反应性[65].
另外,一系列金纳米团簇,如有机可溶性Au25(PET)18-、水溶性Au25(Capt)18-、Au38(SAdm)20、Au25(SR)18等在可见/近红外辐射下可直接敏化,有效促进O2生成1O2[66].
有学者将铁单原子锚定在碳纳米管(Fe—CNT)上形成Fe—C—O配位基序,用于O2活化. 结果表明,此催化剂对O2还原生成H2O2具有优异的活性和选择性[67]. 生成的H2O2作为ROS可用于降解污染物. 此方法对未来的H2O2生产具有较大的潜力.
另外,有学者将钴单原子锚定在多孔N掺杂石墨烯上作为类Fenton催化剂降解双酚A[68]. 虽然其利用了过氧单硫酸盐活化,但为单原子催化剂活化O2提供了参考,也为污染物降解提供了新的思路.
4 活化分子氧用于降解典型新污染物的进展
4.1 典型新污染物分类
新污染物(ECs)的概念于2003年被提出,一般是指环境中未列入常规监测,但能够进入环境并引起生态或人类健康负面影响的人工合成的或天然存在的有机物. 这类污染物通常是已长期存在于环境中,但由于浓度较低,其存在和潜在危害近期才被发现.
新污染物大致分为以下几种类别[69]:药品及个人护理用品(PPCPs)、内分泌干扰物(EDCs)、多氯联苯(PCBs)、多环芳烃(PAHs).
PPCPs作为一种新污染物日益受到人们的关注. PPCPs种类繁杂,包括各类抗生素、人工合成麝香、止痛药、降压药、避孕药、催眠药、减肥药、发胶、染发剂和杀菌剂等. 常见的有四环素、水杨酸、对乙酰氨基酚、恩诺沙星、氯霉素等. 许多PPCPs组分具有较强的生物活性、旋光性和极性,大都以痕量浓度存在于环境中. 它们的主要来源为污水处理厂和日常清洗,可通过饮用水或食物损害人体.
内分泌干扰物EDCs,也称为环境激素,是一种外源性干扰内分泌系统的化学物质. 其多为有机污染物,这类物质会导致动物体和人体生殖器障碍、行为异常、生殖能力下降、幼体死亡. EDCs主要有以下几种类型:农药和除草剂(如甲草胺、草达灭、六六六、六氯苯等)、工业化合物(如多氯联苯、多溴联苯、双酚A、邻苯二甲酸酯类、烷基酚类、硝基苯类等)、类固醇雌激素(如己烷雌酚等)、植物和真菌雌激素、有机重金属(如汞、胂)等.
多氯联苯(PCBs)极难溶于水而易溶于脂肪和有机溶剂,并且很难分解,因此它能够在生物体脂肪中大量富集,造成危害. 根据联苯被氯取代个数,将PCBs分为一氯联苯、二氯联苯、三氯联苯、四氯联苯、五氯联苯、六氯联苯、七氯联苯、八氯联苯、九氯联苯、十氯联苯.
多环芳烃(PAHs)是煤、石油、木材、烟草、有机高分子化合物等有机物不完全燃烧时产生的挥发性碳氢化合物,是重要的环境和食品污染物. 常见的有萘、苯并[α]芘、苯并[α]蒽等. 部分水体中ECs的浓度如表1所示:

表1 部分水体ECs的浓度
4.2 在降解典型新污染物中的应用
目前,ECs的处理技术主要包括活性炭吸附法、膜过滤法、高级氧化技术. 其中,活化碳吸附通过吸附将污染物从水相转移到固相. 其吸附效率主要取决于材料条件、ECs 的性质和环境条件,优点为不会产生副产物、能耗较低,但其在灭活细菌方面不太有效. 膜过滤主要作用机制为通过尺寸排阻、静电作用将ECs吸附到膜表面,生成生物膜层,之后进行生物辐射,同膜的疏水相互作用,去除效率取决于ECs的理化特性、膜的特性和操作条件. 主要缺点为其只改变了ECs 的相位,促进了分离过程,而没有将ECs真正从环境中移出且能耗较高[1]. 高级氧化技术是一种新氧化技术,其主要将外部氧化剂(O3、H2O2、过硫酸盐等)转化为ROS,利用ROS的强氧化性,可以降解几乎所有有机污染物. 其中,O2是绿色、储量丰富且廉价的氧化剂,可被催化转化为ROS(·OH、·O2-、H2O2、1O2)对有机污染物进行氧化降解,相较于活性炭吸附法和膜过滤法具有条件温和、成本低、无二次污染等优点. 近年来,利用O2活化技术降解水中新污染物已逐渐成为热点. 表2~4列出了目前催化活化O2降解新污染物的部分研究.

表2 有关PPCPs的部分研究
4.3 活化分子氧降解新污染物的典型路径和机理
4.3.1Fe2+/四聚磷酸(TPP)降解氯霉素
Fe2+/TPP/空气(Air)体系活化O2产生·OH和·O2-. 余洁等[74]通过自由基捕获实验证实在降解氯霉素(CAP)过程中起主要作用的是·OH,结合降解过程中的脱氯量(10%)、总有机碳去除率(20%)等数据,提出了氯霉素的降解路径,如图5所示,即以首先断裂侧链的C—N键为主.

图5 Fe2+/TPP体系中氯霉素降解路径[74]Fig.5 Proposed degradation pathway of CAP in Fe2+/TPP/ air system[74]

表4 有关PCBs、PAHs的部分研究
4.3.2蒙脱土/亚纳米级零价铜降解阿特拉津

4.3.3CuS4团簇降解四环素
Li等[83]通过对CuS4团簇活化O2降解四环素的研究,提出了四环素的降解路径. 其中,CuS4团簇可通过单电子途径将O2活化为·O2-,部分转化生成H2O2,H2O2进一步反应生成·OH,降解过程如图7所示:在ROS的作用下四环素分子首先发生脱除—NH、—CO、脱水、羟基化反应,之后发生开环反应,并最终被矿化.

图7 CuS4团簇体系中降解四环素路径[83]Fig.7 Proposed degradation pathways of TC by ROS over CuS4 -ZIS[83]

注: RT(retention time)为保留时间.图8 TiO2体系中降解PCB3路径[84]Fig.8 Proposed pathway for the photocatalytic degradation of PCB3 by TiO2[84]
4.3.4TiO2光催化降解4-氯联苯
Zhu等[84]对TiO2体系降解4-氯联苯(PCB3)进行了研究,提出了PCB3的降解路径. 降解过程如图8 所示,·OH取代氯生成4-羟基联苯(主要产物)或发生脱氯反应生成联苯,之后发生开环反应生成,生成小分子醛、醇和酮,最终被氧化为小分子有机酸、CO2.
4.3.5石墨烯-TiO2-Sr( OH)2/ SrCO3纳米复合材料光催化降解菲
Fu等[86]研究了石墨烯-TiO2-Sr( OH)2/ SrCO3体系对于菲的降解,降解路径如图9所示,O2-和OH首先攻击活性高的9或10位点,得到中间体2、3、4、5,之后经进一步氧化降解、烷基化反应,最终被降解为CO2和H2O.

图9 GO-TiO2-Sr(OH)2/SrCO3体系降解菲路径[86]Fig.9 Proposed photocatalytic degradation pathway of phenanthrene by GO-TiO2-Sr(OH)2/SrCO3 under solar irradiation[86]
综上几种典型路径,初步总结出O2活化产生的ROS可攻击有机污染物中较弱的键,发生键的断裂、开环等反应,进而将有机污染物矿化,可为O2活化降解新有机污染物体系提供参考.
5 结语
该研究重点介绍了活化分子氧(O2)降解水中新污染物的基本概念和最新研究进展,包括主要的O2活化机理、主要活性氧物种(ROS)、氧活化策略、新污染物的分类、典型新污染物降解路径,充分展现了O2活化应用于净化水体污染的巨大潜力. 但仍然存在以下问题:
a) 活化O2主要依赖水中的溶氧,O2经由扩散、吸附在催化活性中心上,才能得失电子生成ROS. 由于活性中心位置和数量的限制,其ROS生成效率不高.
b) 活化O2过程中电子利用效率普遍不高,不利于新污染物的高效降解.
c) 针对O2活化技术的相关工艺和设备设计较为少见,不利于未来O2活化技术快速应用于水污染处理.
因此,开发新型催化材料,提高活性中心生成ROS的效率和电子利用效率成为未来有价值的研究方向.
猜你喜欢
中老年保健(2022年3期)2022-08-24
中国矿业(2022年7期)2022-07-15
快乐学习报·教师周刊(2022年3期)2022-04-21
湖南交通科技(2021年4期)2022-01-21
——人-时间资料率比分析与SAS实现
四川精神卫生(2021年4期)2021-09-10
化工设计通讯(2021年4期)2021-01-07
教育周报·教育论坛(2020年3期)2020-10-21
科学(2020年2期)2020-08-24
石油化工(2020年2期)2020-04-04
科技资讯(2018年16期)2018-10-26
