
IL-17信号通路及其在银屑病中的作用
2021-12-21 00:52李欣刘拥军刘广洋
中国医药生物技术 2021年6期
李欣,刘拥军,刘广洋
·综述·
IL-17信号通路及其在银屑病中的作用
李欣,刘拥军,刘广洋
100176 北京贝来生物科技有限公司/北京市亦创生物技术产业研究院干细胞与再生医学研究所
白介素-17(IL-17)是由 T 辅助细胞 Th17 产生的一类炎症因子,可通过直接或间接诱导多种炎症因子、趋化因子介导炎症反应,与银屑病等自身免疫性疾病的发生发展密切相关。银屑病是一种可影响各个年龄段人群的常见的、慢性的、非传染性的炎症性皮肤病,表现为在肘部、膝盖和头皮等部位现清晰的红色鳞片斑块,并且在病变部位易出现慢性-复发性病程[1-2]。银屑病的特征包括表皮的改变(如角质细胞过度增殖和分化),同时伴有明显的炎性浸润和新生血管的生成,皮肤中的浆细胞样树突状细胞、自然杀伤细胞和巨噬细胞产生促炎细胞因子[3],并刺激髓系树突状细胞产生 IL-12 和 IL-23,而 IL-23 可促进 T 细胞分化为 Th17 细胞和 Th22 细胞,其中,Th17 细胞通过产生 IL-17A、IL-17F 等细胞因子,导致了银屑病特有的皮肤炎症,同时 IL-17A 等还参与了正反馈循环,导致银屑病炎症过程反复并导致病情的加重[4-5]。因此,使银屑病的治疗已经从单纯的免疫抑制转向阻断 IL-23/Th-17 途径及通路下游分子,如 IL-17A 及其细胞受体靶点[5]。
本文综述了 IL-17 家族 6 个不同的亚型(IL-17A ~ IL-17F)及其受体,以及 IL-17 信号通路和银屑病相关炎症作用机制,并总结了 IL-17 抑制剂在银屑病治疗的临床应用,为进一步开发治疗银屑病新药提供思路。
1 IL-17 家族及其受体
IL-17 家族共有 6 名成员:IL-17A、IL-17B、IL-17C、IL-17D、IL-17E(又称为 IL-25)和 IL-17F。在 IL-17 家族成员中,IL-17A 与 IL-17F 的同源性最高,约 55%,而其他细胞因子与 IL-17A 只有 16% ~ 30% 的相似度。IL-17 家族中的细胞因子以二硫键结合的同源二聚体形式分泌并发挥作用(除 IL-17B),而 IL-17A 与 IL-17F 也可以形成异源二聚体。Th17 细胞是产生 IL-17 的主要 T 淋巴细胞,该细胞是一种独特的 CD4+T 细胞,通过分泌 IL-17A、IL-17F、IL-21 和 IL-22 等因子,在 T 细胞介导的炎症反应中起着重要作用[6]。除 Th17 外,其他细胞也可以产生 IL-17,如 CD8+T 细胞、γδT 细胞、CD3+CD4-CD8-(双阴性)T 细胞、肥大细胞、自然杀伤细胞、巨噬细胞等[7-8]。
1.1 IL-17 因子家族成员及作用
1.1.1 IL-17A 和 IL-17F IL-17A是最早发现的 IL-17 因子,最初命名为 CTLA-8,1993 年科学家首次从活化的小鼠 T 淋巴细胞中发现。人 IL-17A 是由 155 个氨基酸组成的 30 ~ 35 kD 的蛋白,与小鼠 IL-17A 的同源性高达 60%[9]。
IL-17F 是与 IL-17A 同源性最高的细胞因子,约有 55% 的同源性,均由包括 Th17 细胞、γδT 细胞等在内的 CD4+T 淋巴细胞表达并均受相同基因的调控。IL-17A 和 IL-17F可以以同源二聚体或IL-17A/IL-17F 异源二聚体的形式由免疫细胞分泌,通过 IL17RA 和 IL-17RC 亚基组成的受体二聚体传递刺激信号。IL-17F 在体外对效应细胞的刺激作用与 IL-17A 相似,但作用效力较弱,而 IL-17A 与 IL-17F异源二聚体则可显著增强对效应细胞的作用[10];当 IL-17A、IL-17F 两者共同作用后,可以调节机体炎症反应,并且这种调节作用强于单独使用 IL-17A[11]。因此,在银屑病等皮肤炎症疾病治疗中,相比于 IL-17A,IL-17F 可能是更有潜力的靶点。
1.1.2 IL-17B 和 IL-17D IL-17B 以非共价二聚体的形式分泌,并通过一种含有 IL-17RB 亚基的受体传递信号。IL-17B 在中性粒细胞、神经元和基质细胞以及肠上皮细胞中表达,而被激活的 T 细胞不产生 IL-17B[12]。IL-17B 在关节炎中可能起着致病作用[13-14],在胶原诱导的关节炎小鼠模型中,炎症软骨部位 IL-17B 过度表达,诱导 IL-8 产生进而在软骨部位募集了大量的中性粒细胞,通过 IL-17B 为靶点的中和抗体治疗后,可以改善关节炎小鼠的体征和症状[13],因此,IL-17B 在炎性关节炎的发病机制中起着重要作用。
IL-17D 以二硫键连接的同源二聚体的形式分泌,并通过一种未知的受体发出信号,IL-17D 存在于多种组织中,包括骨骼肌、脑、肺等,可以调节内皮细胞产生细胞因子,如 IL-6、IL-8 和粒细胞-巨噬细胞集落刺激因子(GM-CSF),在控制病毒感染和癌症方面发挥作用[15]。
1.1.3 IL-17C 和 IL-17E(IL-25) IL-17C 与 IL-17A 的同源性只有 23%,与 IL-17A 不同的是,IL-17C 主要由上皮细胞表达,而不是免疫细胞。多项实验发现,IL-17C 与皮肤炎症有关,IL-17C 在银屑病患者[16]和特应性皮炎患者[17]的皮损部位中过表达,体外培养的上皮细胞在炎性因子(如 TNF-α、TLRs)的刺激下可以分泌 IL-17C[18-19],皮肤和肠道的上皮细胞是 IL-17C 的主要靶点,IL-17C 的作用类似于 IL-17A,如促进促炎因子、趋化因子和抗菌肽的生成[16]。小鼠模型实验结果证实了 IL-17C 参与了皮肤炎症过程,银屑病模型小鼠的皮损部位中 IL-17C 表达上调[17],IL-17C 基因敲除的小鼠在咪喹莫特的作用后,与正常小鼠相比皮肤炎症较轻[20];相反,正常小鼠皮内注射过量 IL-17C 后,会出现表皮增厚及显著的银屑病样皮肤炎症[21]。
IL-17E 与 IL-17A 的同源性只有 16%,以二硫键连接的同源二聚体的形式分泌[22],通过与 IL-17RB和 IL-17RA 组成的异二聚体受体复合物结合发挥作用。IL-17E 由多种细胞类型产生,包括上皮细胞、内皮细胞和多种免疫细胞(如 T 细胞、巨噬细胞、II 型髓样细胞、DC、嗜酸性粒细胞和 ILC2)[23]。多项研究结果发现,IL-17E 在几种皮肤炎性疾病的皮损组织中表达上调,如特应性皮炎[24-25]、银屑病[26-27]以及接触性皮炎[28]。在银屑病中,皮损部位表皮角质形成细胞产生的 IL-17E 可以激活巨噬细胞产生炎性细胞因子(TNF、IL-8 等),皮损部位 IL-17E 的表达与中性粒细胞的数量相关,而与 T 细胞的数量呈负相关,表明 IL-17E 可能在皮肤天然免疫过程中发挥作用[26];在体外实验中,IL-17E 可以促进人皮肤成纤维细胞表达促炎细胞因子[13],如 IL-8、CCL-5 等,这些结果表明 IL-17E 可能是银屑病的致病因子[29]。
1.2 IL-17 受体及功能
1995 年科学家发现,IL-17 受体(IL-17 receptor,IL-17R)并不属于任何已知的受体类别,该受体共有 5 个同源亚基,故将这些受体命名为 IL-17RA、IL-17RB、IL-17RC、IL-17RD 和 IL-17RE,并归类为一类新的受体——IL-17R 家族[30]。IL-17 受体家族的 5 个成员均有保守的结构特征,其中 IL-17RA 是研究最多的受体,可以与IL-17RB、IL-17RC 和 IL-17RE 形成异源二聚体复合物。而 IL-17 细胞因子都通过结合这些不同亚基组成的异源二聚体受体而发挥作用:IL-17A 同源二聚体、IL-17F 同源二聚体、IL-17A/F异源二聚体通过含有 IL-17RA 和 IL-17RC 的异源二聚体传递信号;IL-17C 信号通路通过 IL-17RA/RE 复合物介导宿主防御,与 IL-17A 一样,参与自身免疫性疾病的发病机制[21];IL-17E(IL-25)通过含有 IL-17RA 和 IL-17RB 的异构体受体传递信号,IL-17B 通过 IL-17RB 发出信号[31]。
1.3 IL-17 作用靶点
IL-17A 作用于多种细胞类型,包括内皮细胞、成纤维细胞、软骨细胞、滑膜细胞、单核细胞和上皮细胞(包括角质形成细胞)。IL-17A 和 IL-17F 直接作用于角质形成细胞,刺激银屑病皮损组织中的分子大量分泌,例如细胞因子、β-防御素、抗菌肽(AMPs)以及刺激中性粒细胞、巨噬细胞和淋巴细胞分泌大量的趋化因子,如 IL-8、CCL-20 和 CCL-2[32]。IL-17C 类似于 IL-17A 和 IL-17F,IL-17C 也可以作用于角质形成细胞,诱导产生 β-防御素(hBD2)和粒细胞集落刺激因子(G-CSF)。IL-17E 与 IL-17A、IL-17C 和 IL-17F 的不同之处在于,它通常在过敏反应过程中由上皮细胞产生,并诱导 Th2 型反应[33]。在小鼠银屑病模型中,与非皮损皮肤相比,皮损部位中 IL-17E、IL-17B 和 IL-17D的表达没有增加。血清学结果显示,模型组小鼠血清中 Th17 细胞表达的细胞因子表达量增加,而治疗组血清中的 IL-17A 和 IL-17F 表达量降低[34]。
2 IL-17 依赖的炎症和信号转导
2.1 IL-17A 与其他分子协同增强炎症反应
研究发现,IL-17A 介导的 NF-κB 信号通路的激活依赖于 TRAF-6,在 Act1 与受体复合物结合后,TRAF-6 通过与 Act1 的 TRAF 结合端的相互作用而被招募,Act1 的 U 型结构域作为一种 E3 泛素连接酶促进其目标蛋白的 Lys63 连接泛素化,进而促进后续的蛋白质-蛋白质相互作用[35]。在 IL-17A 刺激下,泛素化的 TRAF-6 在野生型细胞中可以检测到,但在缺乏 Act1 的细胞中检测不到,而 Act1 的 U 型结构的缺失会阻止 Act1 介导的 TRAF-6 的泛素化,并削弱 IL-17 依赖的反应。泛素化的 TRAF-6 进一步激活下游依赖 TRAF-6 的转化生长因子 β 激活激酶 1(TAK1),进而激活 NF-κB 通路[36]。
单独的 IL-17A 是一种功能较弱的 NF-κB 激活剂,但它之所以成为重要的致病因子,是因为 IL-17A 可以与其他细胞因子(例如 TNF-α)协同作用,促进和延长炎症反应,TNF-α 是一种作用力强的 NF-κB 激活剂,可以诱导高度不稳定的促炎因子 mRNA 的表达,而 IL-17A 可以通过稳定这些 mRNA 来增强趋化因子的表达。这一机制涉及另外两个 TRAF 分子:TRAF-2 和 TRAF-5,以及一种名为 IKKi(也称为 IKKξ)的激酶,IKKi 在 IL-17A 刺激下被招募到 IL-17R-Act1 复合物中,进而招募 TRAF-2 和 TRAF-5,但不招募 TRAF-6,形成 Act1/TRAF-2/TRAF-5 复合体,增强了 CXCL1 mRNA 的稳定性[37-38],TRAF-2 和 TRAF-5 通过激活的 IKKi 的作用参与 IL-17A 依赖的基因稳定性。IKKi 缺乏可以抑制 Act1/TRAF-2/TRAF-5 复合物的形成,并导致 mRNA 稳定表达功能的丧失,但不影响 Act1-TRAF-6-NF-κB 的活性。在 IL-17A 的刺激下,小鼠胚胎成纤维细胞中 TRAF-6 的缺失导致 NF-κB 活性的丧失及 IL-6 的产生,而依赖 IL-17A 的 mRNA 的稳定性没有受到影响[39],因此,IL-17 通过 TRAF-6 非依赖性途径的 mRNA 稳定表达在调节促炎细胞因子和趋化因子的协同表达中起重要作用。
2.2 IL-17A 信号的调控
IL-17A 的刺激导致 TRAF-4 向 IL-17R 复合体募集,TRAF-4 是 IL-17 信号负调控因子,利用与 Act1 上 TRAF-6 相同的 TRAF 结合位点,与 TRAF6 竞争 Act1 结合,TRAF-4 缺失小鼠表现出依赖 IL-17 信号的细胞因子的表达明显增多[40]。TRAF-3 也被证实是 IL-17 信号级联反应中一个重要的负调控因子,直接与 IL-17R 结合,并干扰 IL-17R-Act1-TRAF-6 复合物的形成,TRAF-3 基因的敲除促进了 NF-κB 和丝裂原活化蛋白激酶(MAPK)的激活,并增强了 IL-17A 依赖的促炎基因的表达,在 IL-17 刺激下,C/EBPβ 在 ERK 和糖原合成酶激酶 3β(GSK3β)的作用下发生磷酸化,而 C/EBPβ 上的这些磷酸化分子抑制 IL-17 依赖的促炎基因的表达[41]。
最近的研究发现,一种脱泛素酶 USP25 可以抑制 IL-17R 信号转导[42],体外实验发现,IL-17A 处理USP25 缺陷细胞后,导致缺陷细胞中 TRAF-5 和 TRAF-6 的超泛素化,延长了编码 CXCL1 mRNA 的半衰期,增强了 JNK 的磷酸化,因此,USP25 缺乏会导致 IL-17A 依赖的趋化因子和细胞因子产生过度,在体内实验中,USP25 缺陷小鼠表现出 IL-17 介导的肺部炎症增强。这些实验表明,USP25 可能通过抑制 TRAF-5 和 TRAF-6 的泛素化来调节 IL-17 信号;另一种去泛素酶 A20 是由 TNF-α 诱导蛋白 3(TNFAIP3)编码的一种肿瘤抑制因子,直接与 IL-17RA 的远端结构域相互作用,并以 TRAF-6 以 IL-17依赖的方式结合,以限制依赖 IL-17 的 NF-κB 和 MAPK 的激活[43]。
IL-17 依赖的 NF-κB 激活也可以导致人成纤维样滑膜细胞、小鼠原代肾细胞和星形胶质细胞中 RNA miR-23b 的下调[44],miR-23b 通过靶向 Tab2、Tab3 和 IKKα 等上游信号介质抑制 NF-κB 活性和炎性细胞因子的表达[44-45]。
3 IL-17 家族在银屑病发病过程的作用及机制
大量研究数据证明,IL-17 和 Th17 的相关细胞因子,在银屑病发病过程中起到重要作用。将 IL-17 家族中多个相关基因与银屑病的发病机制联系在一起(表 1),对银屑病在分子与细胞水平的分析提高了我们对疾病发病机制的了解。
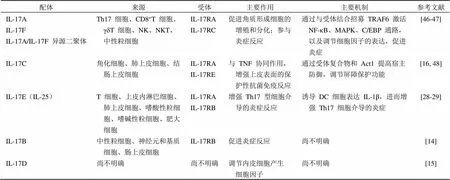
表 1 IL-17 家族及其受体的作用及机制
多项临床前试验结果证实了 IL-17 在银屑病中的作用,包括 IL-17 通路的变化以及 IL-17A 和IL-17RA 的抑制作用,均与银屑病成功治疗有一定的相关性。与健康皮肤相比,银屑病皮损部位中 CD4+Th17 细胞数量增多,并且这些 Th17 细胞具有分化产生 IFN-γ 的能力,对皮损样本进行免疫染色,结果显示 IL-17A 阳性的细胞数增多,经过治疗后,皮损部位的 CD4+Th17 细胞数量减少[49]。在之后的实验中发现,IL-17 阳性的细胞可能是 CD8+T 细胞(Tc17)、淋巴组织诱导细胞(LTi)、肥大细胞和(或)中性粒细胞,这些不同类型的细胞在银屑病发病机制中有着非常重要的作用[50]。
全基因组关联研究发现,银屑病患者基因组中存在一系列单核苷酸多态性(SNPs),将 IL-17 细胞因子与银屑病易感性联系起来[51]。同样,银屑病小鼠模型显示了 Th17 细胞因子在介导皮肤炎症发病机制中的重要性。在小鼠皮内注射 IL-23 可以导致皮肤增生,而 IL-17RA 缺乏、IL-17A 缺乏或抗 IL-17 抗体中和可以减轻这种状况[52]。在银屑病患者皮损部位和血液中,Th17 细胞和 IL-17(IL-17A、IL-17C)水平均升高,且与病情严重程度具有相关性,对银屑病进行成功治疗后,患者血清中 IL-17 水平也有所下降。以上研究进一步证实 IL-17 信号通路在银屑病发病过程中有着重要的作用。
4 IL-17 信号通路在银屑病病理过程的机制研究
IL-17 信号通路在银屑病病理过程中发挥关键作用。IL-17A/F 可激活角质形成细胞产生细胞因子和白细胞介素,如 IL-8,而 IL-8 对中性粒细胞有着很强的趋化作用,同时,IL-17A/F 可上调角质形成细胞表达的趋化因子(如 CXCL-1、CXCL-3、CXLC-5、CXCL-6),这一过程也与中性粒细胞的作用有关。在银屑病中,IL-17A 还诱导角质形成细胞表达 CCL-20,向皮肤募集更多的 Th17 细胞和树突状细胞[53]。此外,IL-17A 可以上调抗菌肽(如β-防御素)[54],刺激树突状细胞和成纤维细胞产生 IL-6,进一步促进 T 细胞向 Th17 分化[55],IL-17 通过刺激成纤维细胞产生血管内皮生长因子来促进内皮细胞的增殖和血管生成。
Act1 可能在 IL-17 信号传导中发挥重要作用(图 1);Act1 是一种直接与 IL-17R 复合物结合的胞内蛋白,可以激活 IL-17R 并启动两条不同的信号转导通路。当 IL-17 与 IL-17R 结合时,Act1 与 IL-17R 复合物结合,激活下游通路[56],Act1 的招募是 IL-17 信号通路的一个标志,这一过程在其他任何已知的受体类别中都不存在,Act1 可以激活多条独立的信号通路,作为不同 TRAF 蛋白的对接站,当 TRAF-6 和它的泛素化(Act1 作为 E3 泛素连接酶)被招募时,一系列的分子作用被开启,导致 NF-κB 基因的激活[35, 57]。NF-κB 信号通路可以通过与 TRAF-6 的相互作用来启动,在 TRAF-6 非依赖途径中,Act1 可以与TRAF-2/5、剪接调节因子 SRSF1 结合成复合体,促进下游炎症因子 mRNA 的稳定表达[38],表明银屑病可能通过 Act1-TRAF-5-mRNA 途径促进炎性因子的表达(图 1)。
在 IL-23 的刺激下,幼稚 T 细胞与激活的树突状细胞(DC)相互作用后分化为 Th17,Th17 细胞分泌 IL-17(包括 IL-17A 和 IL-17F),与角质形成细胞的 IL-17R 结合,角质形成细胞在 IL-17 的刺激下异常增殖和分化,产生趋化因子和血管生成因子,刺激炎症细胞的聚集,建立正反馈循环。趋化因子 CCL20 是角质形成细胞衍生的重要核心成分,其功能是将 Th1、Th22 和 Th17 细胞召集到活跃的受损皮肤中,Th1 和 Th22 细胞通过产生炎症因子来放大核心反应,以及角质形成细胞衍生的血管生成因子和趋化因子,最终导致银屑病皮损的形成(图 2)。
5 IL-17 抑制剂在银屑病治疗的临床应用
随着我们对银屑病病理生理学的不断了解,银屑病的治疗方法在过去 20 年里有了巨大的进步,大量的生物制剂特别是单克隆抗体药物被 FDA 批准上市治疗中、重度银屑病,这些抗体药物均针对特定的靶点,通过与致炎致病性抗原或者细胞因子的结合,特异性地阻断其余受体的结合,阻断炎症因子信号传递,进而阻断皮肤炎症的发展,实现治疗银屑病的目的,这类抗体药物在临床应用有良好的治疗效果及较少的不良反应。这类药物主要包括 TNF-α、IL-17、IL-12/23 和 IL-23 等拮抗剂。其中,通过单克隆抗体中和 IL-17A(secukinumab 和 ixekizumab)或 IL-17RA(brodalumab)是治疗银屑病非常有效的方法[58-59],这三种抗体类药物的共同适应证为中、重度斑块型银屑病。其中 secukinumab 及 ixekizumab 通过皮下注射的方式进行治疗,12 周后,分别有 77% 和 83% 的患者达到 PASI-75,治疗效果优于依那西普(TNF-α 拮抗剂)[58, 60],在剂量使用方面,ixekizumab 的使用剂量低于 secukinumab,安全性也优于 secukinumab[61]。而 brodalumab 是 2017 年经 FDA 批准上市,适应证更为局限,仅适用于斑块型银屑病,皮下注射的方式进行治疗 12 周后,有 83% 的患者达到了 PASI-75,治疗效果优于 ustekinumab(IL-12/23 拮抗剂)[62]。在这些抗体药物使用过程中,会有一些不良反应,例如:鼻咽炎、头痛和腹泻[63-65]。
除了以上获得许可的药物外,针对多个 IL-17 家族成员其他几个分子(如:bimekizumab 可以双重中和 IL-17A 和 IL-17F[11])或者 IL-17 通路(上游即 IL-23 及下游信号)的药物正在临床开发中[66],因此,更好地了解 IL-17 家族的结构和其成员在炎症方面的作用是至关重要的[67]。
6 小结与展望
银屑病是一种慢性复发性炎症性皮肤病,严重影响患者的生活质量,通过传统治疗如维 A 酸类药物及糖皮质激素的治疗,不能达到良好的治疗效果且有较多的不良反应,而以致病因子作为治疗靶点的生物制剂以其效果良好、不良反应较少的优势在银屑病的治疗中慢慢占据了重要地位。近年来,随着对 IL-17 来源及对信号转导通路的深入研究,我们对 IL-17 家族在疾病中的生物学功能有了更深入的了解,然而,对于 IL-17 在不同疾病中的促炎和组织保护作用的复杂性,目前尚有待进一步明确,首先,更好地理解 IL-17 信号在炎症和自身免疫环境中的作用机制,对于发现新的治疗靶点至关重要,这些靶点将为治疗包括银屑病在内的自身免疫性疾病提供新的潜在治疗方法;其次,以 IL-17 为靶点的生物制剂类药物虽然在临床应用上表现出良好的治疗效果,但由于给药频次高、治疗成本高以及患者的适用性存在局限性等客观条件,影响了生物制剂的使用范围。因此,发现新的治疗靶点或与结合基因转导技术相结合,不断优化更新银屑病的治疗方案,可为银屑病患者提供新的希望。
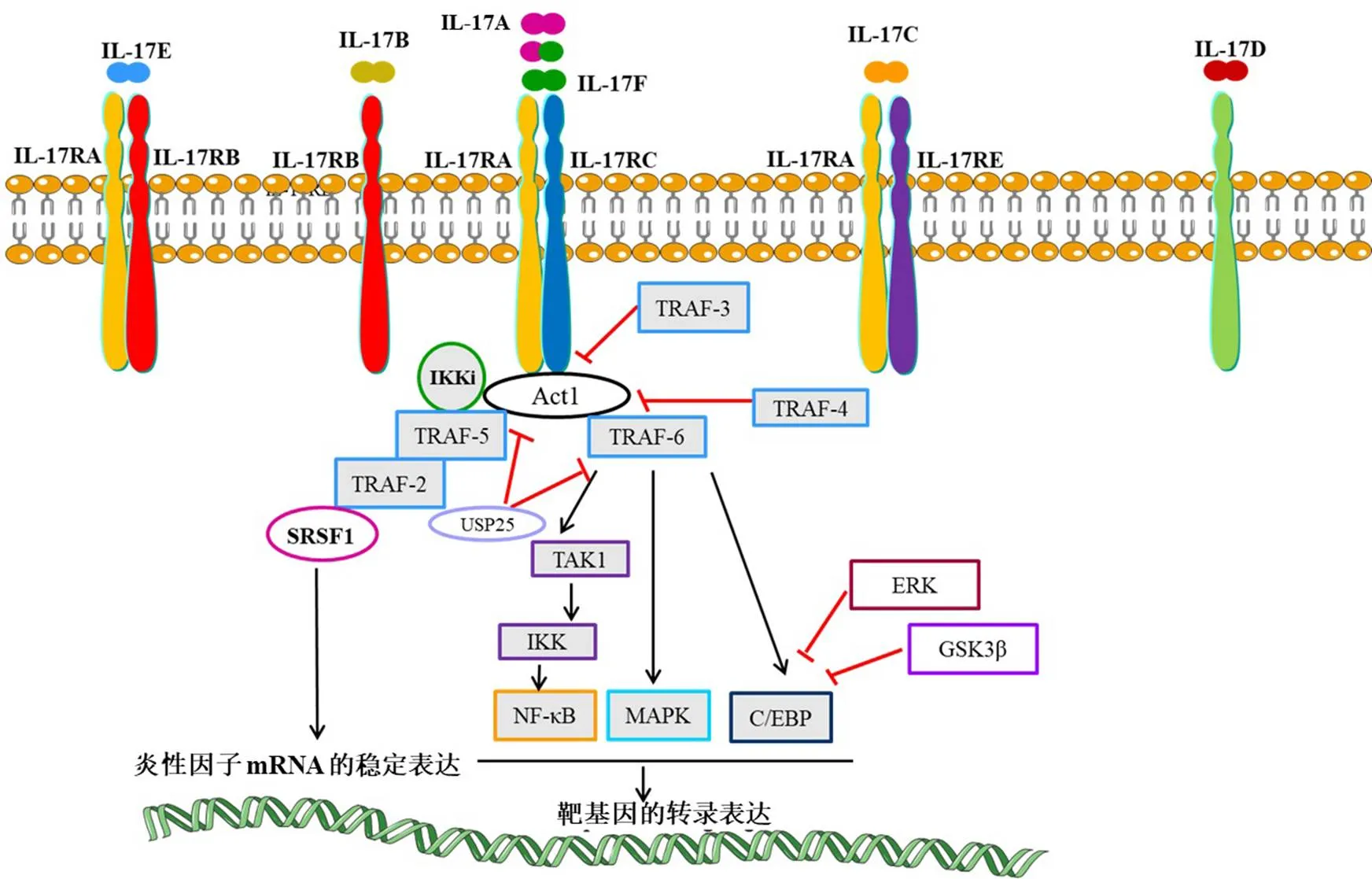
图1 IL-17 细胞因子、受体与信号转导
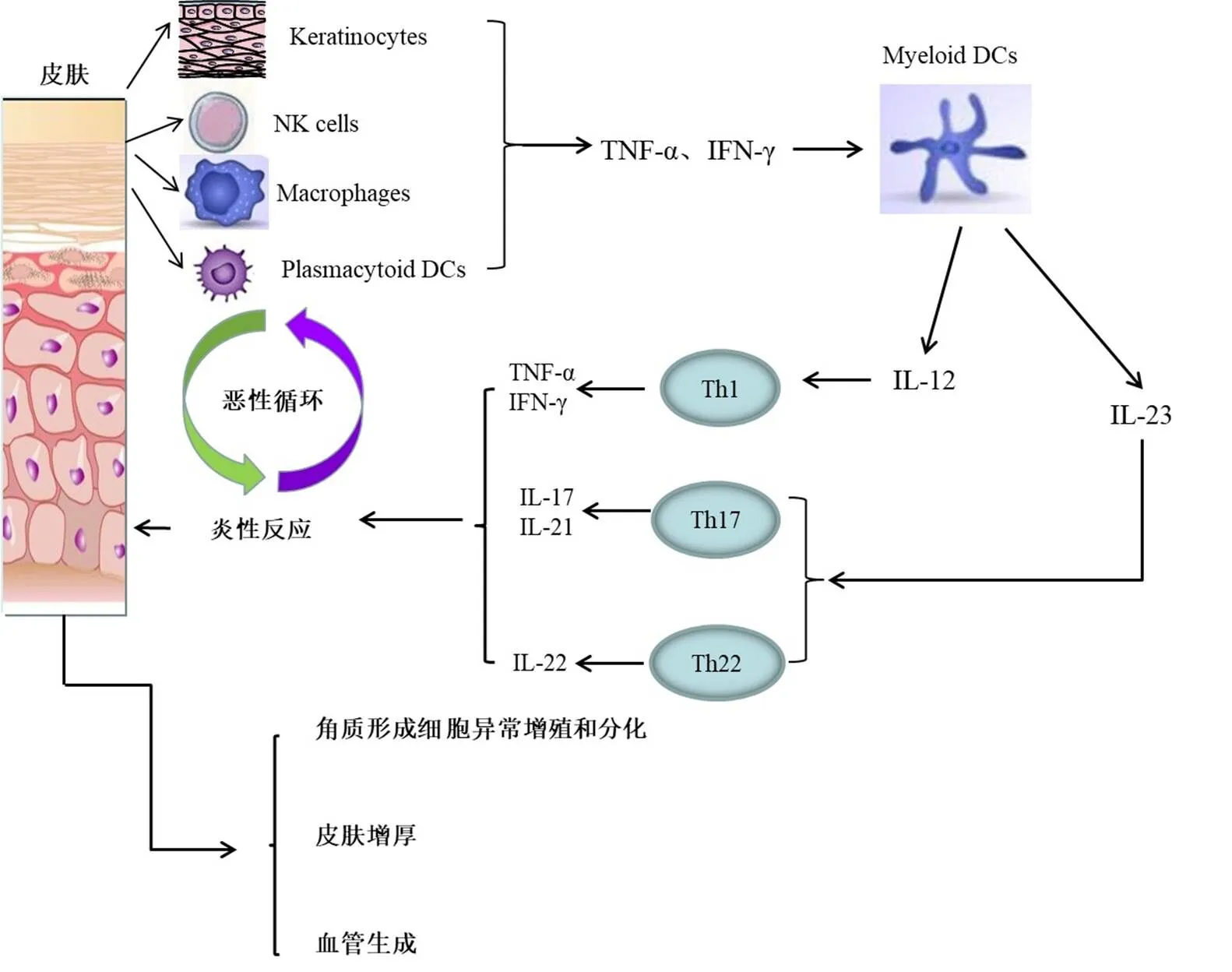
图 2 IL-17 在银屑病发病机制中的作用
[1] Boehncke WH, Schön MP. Psoriasis. Lancet, 2015, 386(9997):983- 994.
[2] Daudén E, Castañeda S, Suárez C, et al. Clinical practice guideline for an integrated approach to comorbidity in patients with psoriasis. J Eur Acad Dermatol Venereol, 2013, 27(11):1387-1404.
[3] Cai Y, Fleming C, Yan J. New insights of T cells in the pathogenesis of psoriasis. Cell Mol Immunol, 2012, 9(4):302-309.
[4] Boehncke WH. Etiology and pathogenesis of psoriasis. Rheum Dis Clin North Am, 2015, 41(4):665-675.
[5] Lynde CW, Poulin Y, Vender R, et al. Interleukin 17A: toward a new understanding of psoriasis pathogenesis. J Am Acad Dermatol, 2014, 71(1):141-150.
[6] Nestle FO, Kaplan DH, Barker J. Psoriasis. N Engl J Med, 2009, 361(5):496-509.
[7] Wilson NJ, Boniface K, Chan JR, et al. Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol, 2007, 8(9):950-957.
[8] Miossec P, Kolls JK. Targeting IL-17 and TH17 cells in chronic inflammation. Nat Rev Drug Discov, 2012, 11(10):763-776.
[9] Fossiez F, Djossou O, Chomarat P, et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J Exp Med, 1996, 183(6):2593-2603.
[10] Amatya N, Garg AV, Gaffen SL. IL-17 signaling: the yin and the yang. Trends Immunol, 2017, 38(5):310-322.
[11] Glatt S, Baeten D, Baker T, et al. Dual IL-17A and IL-17F neutralisation by bimekizumab in psoriatic arthritis: evidence from preclinical experiments and a randomised placebo-controlled clinical trial that IL-17F contributes to human chronic tissue inflammation. Ann Rheum Dis, 2018, 77(4):523-532.
[12] Bie Q, Jin C, Zhang B, et al. IL-17B: a new area of study in the IL-17 family. Mol Immunol, 2017, 90:50-56.
[13] Yamaguchi Y, Fujio K, Shoda H, et al. IL-17B and IL-17C are associated with TNF-alpha production and contribute to the exacerbation of inflammatory arthritis. J Immunol, 2007, 179(10): 7128-7136.
[14] Kouri VP, Olkkonen J, Ainola M, et al. Neutrophils produce interleukin-17B in rheumatoid synovial tissue. Rheumatology (Oxford), 2014, 53(1):39-47.
[15] Seelige R, Washington A Jr, Bui JD. The ancient cytokine IL-17D is regulated by Nrf2 and mediates tumor and virus surveillance. Cytokine, 2017, 91:10-12.
[16] Johnston A, Fritz Y, Dawes SM, et al. Keratinocyte overexpression of IL-17C promotes psoriasiform skin inflammation. J Immunol, 2013, 190(5):2252-2262.
[17] Vandeghinste N, Klattig J, Jagerschmidt C, et al. Neutralization of IL-17C reduces skin inflammation in mouse models of psoriasis and atopic dermatitis. J Invest Dermatol, 2018, 138(7):1555-1563.
[18] Johansen C, Vinter H, Soegaard-Madsen L, et al. Preferential inhibition of the mRNA expression of p38 mitogen-activated protein kinase regulated cytokines in psoriatic skin by anti-TNFalpha therapy. Br J Dermatol, 2010, 163(6):1194-1204.
[19] Roth SA, Simanski M, Rademacher F, et al. The pattern recognition receptor NOD2 mediates Staphylococcus aureus-induced IL-17C expression in keratinocytes. J Invest Dermatol, 2014, 134(2):374-380.
[20] Monin L, Gudjonsson JE, Childs EE, et al. MCPIP1/Regnase-1 restricts IL-17A- and IL-17C-dependent skin inflammation. J Immunol, 2017, 198(2):767-775.
[21] Ramirez-Carrozzi V, Sambandam A, Luis E, et al. IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat Immunol, 2011, 12(12):1159-1166.
[22] Gaffen SL. Recent advances in the IL-17 cytokine family. Curr Opin Immunol, 2011, 23(5):613-619.
[23] Xu M, Dong C. IL-25 in allergic inflammation. Immunol Rev, 2017, 278(1):185-191.
[24] Deleuran M, Hvid M, Kemp K, et al. IL-25 induces both inflammation and skin barrier dysfunction in atopic dermatitis. Chem Immunol Allergy, 2012, 96:45-49.
[25] Aktar MK, Kido-Nakahara M, Furue M, et al. Mutual upregulation of endothelin-1 and IL-25 in atopic dermatitis. Allergy, 2015, 70(7): 846-854.
[26] Senra L, Stalder R, Alvarez Martinez D, et al. Keratinocyte-derived IL-17E contributes to inflammation in psoriasis. J Invest Dermatol, 2016, 136(10):1970-1980.
[27] Kim BE, Bin L, Ye YM, et al. IL-25 enhances HSV-1 replication by inhibiting filaggrin expression, and acts synergistically with Th2 cytokines to enhance HSV-1 replication. J Invest Dermatol, 2013, 133(12):2678-2685.
[28] Suto H, Nambu A, Morita H, et al. IL-25 enhances TH17 cell-mediated contact dermatitis by promoting IL-1beta production by dermal dendritic cells. J Allergy Clin Immunol, 2018, 142(5):1500- 1509, e10.
[29] Batalla A, Coto E, González-Lara L, et al. Association between single nucleotide polymorphisms IL17RA rs4819554 and IL17E rs79877597 and Psoriasis in a Spanish cohort. J Dermatol Sci, 2015, 80(2):111- 115.
[30] Moseley TA, Haudenschild DR, Rose L, et al. Interleukin-17 family and IL-17 receptors. Cytokine Growth Factor Rev, 2003, 14(2):155- 174.
[31] Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol, 2009, 9(8):556-567.
[32] Chiricozzi A, Guttman-Yassky E, Suarez-Farinas M, et al. Integrative responses to IL-17 and TNF-alpha in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J Invest Dermatol, 2011, 131(3):677-687.
[33] Barlow JL, Mckenzie AN. IL-25: a key requirement for the regulation of type-2 immunity. Biofactors, 2009, 35(2):178-182.
[34] Nakajima K, Kanda T, Takaishi M, et al. Distinct roles of IL-23 and IL-17 in the development of psoriasis-like lesions in a mouse model.J Immunol, 2011, 186(7):4481-4489.
[35] Liu C, Qian W, Qian Y, et al. Act1, a U-box E3 ubiquitin ligase for IL-17 signaling. Sci Signal, 2009, 2(92):ra63.
[36] Gu C, Wu L, Li X. IL-17 family: cytokines, receptors and signaling. Cytokine, 2013, 64(2):477-485.
[37] Bulek K, Liu C, Swaidani S, et al. The inducible kinase IKKi is required for IL-17-dependent signaling associated with neutrophilia and pulmonary inflammation. Nat Immunol, 2011, 12(9):844-852.
[38] Sun D, Novotny M, Bulek K, et al. Treatment with IL-17 prolongs the half-life of chemokine CXCL1 mRNA via the adaptor TRAF5 and the splicing-regulatory factor SF2 (ASF). Nat Immunol, 2011, 12(9):853- 860.
[39] Hartupee J, Liu C, Novotny M, et al. IL-17 signaling for mRNA stabilization does not require TNF receptor-associated factor 6.J Immunol, 2009, 182(3):1660-1666.
[40] Zepp JA, Liu C, Qian W, et al. Cutting edge: TNF receptor-associated factor 4 restricts IL-17-mediated pathology and signaling processes.J Immunol, 2012, 189(1):33-37.
[41] Shen F, Li N, Gade P, et al. IL-17 receptor signaling inhibits C/EBPbeta by sequential phosphorylation of the regulatory 2 domain. Sci Signal, 2009, 2(59):ra8.
[42] Zhong B, Liu X, Wang X, et al. Negative regulation of IL-17-mediated signaling and inflammation by the ubiquitin-specific protease USP25. Nat Immunol, 2012, 13(11):1110-1117.
[43] Garg AV, Ahmed M, Vallejo AN, et al. The deubiquitinase A20 mediates feedback inhibition of interleukin-17 receptor signaling. Sci Signal, 2013, 6(278):ra44.
[44] Zhu S, Pan W, Song X, et al. The microRNA miR-23b suppresses IL-17-associated autoimmune inflammation by targeting TAB2, TAB3 and IKK-α. Nat Med, 2012, 18(7):1077-1086.
[45] Hu R, O'connell RM. MiR-23b is a safeguard against autoimmunity. Nat Med, 2012, 18(7):1009-1010.
[46] Herjan T, Yao P, Qian W, et al. HuR is required for IL-17-induced Act1-mediated CXCL1 and CXCL5 mRNA stabilization. J Immunol, 2013, 191(2):640-649.
[47] Ramani K, Garg AV, Jawale CV, et al. The kallikrein-kinin system: A novel mediator of IL-17-driven anti-candida immunity in the kidney. PLoS Pathog, 2016, 12(11):e1005952.
[48] Whibley N, Tritto E, Traggiai E, et al. Antibody blockade of IL-17 family cytokines in immunity to acute murine oral mucosal candidiasis. J Leukoc Biol, 2016, 99(6):1153-1164.
[49] Kryczek I, Bruce AT, Gudjonsson JE, et al. Induction of IL-17+ T cell trafficking and development by IFN-gamma: mechanism and pathological relevance in psoriasis. J Immunol, 2008, 181(7):4733- 4741.
[50] Lin AM, Rubin CJ, Khandpur R, et al. Mast cells and neutrophils release IL-17 through extracellular trap formation in psoriasis.J Immunol, 2011, 187(1):490-500.
[51] Perera GK, Di Meglio P, Nestle FO. Psoriasis. Annu Rev Pathol, 2012, 7:385-422.
[52] Rizzo HL, Kagami S, Phillips KG, et al. IL-23-mediated psoriasis-like epidermal hyperplasia is dependent on IL-17A. J Immunol, 2011, 186(3):1495-1502.
[53] Nograles KE, Zaba LC, Guttman-Yassky E, et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br J Dermatol, 2008, 159(5):1092- 1102.
[54] Conti HR, Gaffen SL. IL-17-mediated immunity to the opportunistic fungal pathogen candida albicans. J Immunol, 2015, 195(3):780-788.
[55] Carrier Y, Ma HL, Ramon HE, et al. Inter-regulation of Th17 cytokines and the IL-36 cytokines in vitro and in vivo: implications in psoriasis pathogenesis. J Invest Dermatol, 2011, 131(12):2428-2437.
[56] Ellinghaus E, Ellinghaus D, Stuart PE, et al. Genome-wide association study identifies a psoriasis susceptibility locus at TRAF3IP2. Nat Genet, 2010, 42(11):991-995.
[57] Song X, Qian Y. The activation and regulation of IL-17 receptor mediated signaling. Cytokine, 2013, 62(2):175-182.
[58] Griffiths CE, Reich K, Lebwohl M, et al. Comparison of ixekizumab with etanercept or placebo in moderate-to-severe psoriasis (UNCOVER-2 and UNCOVER-3): results from two phase 3 randomised trials. Lancet, 2015, 386(9993):541-551.
[59] Papp KA, Langley RG, Sigurgeirsson B, et al. Efficacy and safety of secukinumab in the treatment of moderate-to-severe plaque psoriasis: a randomized, double-blind, placebo-controlled phase II dose-ranging study. Br J Dermatol, 2013, 168(2):412-421.
[60] Langley RG, Elewski BE, Lebwohl M, et al. Secukinumab in plaque psoriasis--results of two phase 3 trials. N Engl J Med, 2014, 371(4): 326-338.
[61] Wang J, Bhatia A, Krugliak Cleveland N, et al. Rapid onset of inflammatory bowel disease after receiving secukinumab infusion. ACG Case Rep J, 2018, 5:e56.
[62] Bauer E, Lucier J, Furst DE. Brodalumab -an IL-17RA monoclonal antibody for psoriasis and psoriatic arthritis. Expert Opin Biol Ther, 2015, 15(6):883-893.
[63] Kamata M, Tada Y. Safety of biologics in psoriasis. J Dermatol, 2018, 45(3):279-286.
[64] Bonomo L, Ghoneim S, Levitt J. A case of granuloma annulare associated with secukinumab use. Case Rep Dermatol Med, 2017, 2017:5918708.
[65] Mease PJ, van der Heijde D, Ritchlin CT, et al. Ixekizumab, an interleukin-17A specific monoclonal antibody, for the treatment of biologic-naive patients with active psoriatic arthritis: results from the 24-week randomised, double-blind, placebo-controlled and active (adalimumab)-controlled period of the phase III trial SPIRIT-P1. Ann Rheum Dis, 2017, 76(1):79-87.
[66] Conrad C, Gilliet M. Psoriasis: from pathogenesis to targeted therapies. Clin Rev Allergy Immunol, 2018, 54(1):102-113.
[67] Brembilla NC, Senra L, Boehncke WH. The IL-17 family of cytokines in psoriasis: IL-17A and beyond. Front Immunol, 2018, 9:1682.
10.3969/j.issn.1673-713X.2021.06.009
北京市科技计划课题(Z211100002521006)
刘广洋,Email:liugy04@163.com
2021-09-02
猜你喜欢
世界中医药(2022年22期)2022-12-14
昆明医科大学学报(2021年12期)2021-12-30
天津医科大学学报(2021年4期)2021-08-21
现代临床医学(2021年4期)2021-07-31
皮肤病与性病(2021年3期)2021-07-30
现代仪器与医疗(2021年2期)2021-07-21
昆明医科大学学报(2021年1期)2021-02-07
心肺血管病杂志(2020年5期)2021-01-14
健康之家(2016年10期)2016-10-28
医学研究杂志(2015年12期)2015-06-10
