
高锰酸钾氧化-硫酸亚铁铵滴定法测定碳化钒中钒的含量
2021-12-16 08:40王录锋王彬彬谢秀琼先小容张高庆李国伟
理化检验-化学分册 2021年12期
王录锋,王彬彬,谢秀琼,先小容,张高庆,李国伟
(1.攀枝花学院 钒钛学院,攀枝花 617000;2.国家钒钛制品质量监督检验中心,攀枝花 617000)
碳化钒是一种重要的合金添加剂,能提高硬质合金的耐蚀性、耐磨性、强度等综合性能,因此广泛应用于合金中[1]。碳化钒中一般钒的质量分数为75%~83%,碳的质量分数为9%~14%[2]。
目前,高含量钒的测定方法主要有氧化还原滴定法[3-5]、电感耦合等离子体原子发射光谱法(ICPAES)[6-9]、X 射线荧光光谱法[10-11]和分光光度法[12]。ICP-AES不宜用于分析钒的质量分数大于45%的样品;X 射线荧光光谱法操作简单、分析快速,但仪器昂贵;分光光度法操作简单,但分析效率低;化学滴定法由于准确度高、易于掌握、操作成本低等优点,目前被广泛运用。钒的滴定法根据使用的氧化剂可分为过硫酸铵氧化-硫酸亚铁铵滴定法、高锰酸钾氧化-硫酸亚铁铵滴定法、高氯酸氧化-硫酸亚铁铵滴定法[13-14],以前两种为主,后者应用较少。高锰酸钾氧化-硫酸亚铁铵滴定法只需在常温下就可将钒氧化,其中过量的高锰酸钾在常温下也可采用亚硝酸钠还原;而过硫酸铵氧化-硫酸亚铁铵滴定法则需在加热条件下才能氧化钒,其中过量的过硫酸铵在煮沸条件下才能使其完全破坏掉,且需冷却至常温后才可进行下一步,操作相对更复杂。本工作通过优化碳化钒中钒的测定条件及方法,建立了高锰酸钾氧化-硫酸亚铁铵滴定法测定碳化钒中钒含量的方法,结果令人满意。
1 试验部分
1.1 仪器与试剂
X′Pert3 Powder型X 射线衍射仪;SA224SCW 型电子天平;TM-0914型陶瓷纤维马弗炉。
钒标准溶液:4.0 g·L-1,称取0.7142 g预先经105 ℃烘1 h并置于干燥器内冷却至室温的五氧化二钒于200 mL烧杯中,加入50%(体积分数,下同)硫酸溶液10 mL后加热,冷却后再加水30 mL,再次冷却后转移至100 mL 容量瓶中,以水稀释至刻度,混匀。
重铬酸钾标准溶液:0.07000 mol·L-1(以1/6 K2Cr2O7计),称取3.4322g预先经120℃烘1h干燥至恒重后置于干燥器中冷却的重铬酸钾基准试剂于250 mL 烧杯中,用水溶解后转移至1 L 容量瓶中,用水稀释至刻度,混匀。
硫酸亚铁铵标准滴定溶液:0.10 mol·L-1,称取39.2131g硫酸亚铁铵[(NH4)2Fe(SO4)2·6H2O]溶于5%(体积分数,下同)硫酸溶液中,并用5%硫酸溶液稀释至1 L,混匀。
硫酸亚铁铵溶液:50 g·L-1,称取5 g硫酸亚铁铵溶于100 mL的5%硫酸溶液中,混匀。
N-苯代邻氨基苯甲酸溶液:2 g·L-1,称取0.2 gN-苯代邻氨基苯甲酸溶于少量水中,加0.2 g无水碳酸钠后低温加热溶解,用水稀释至100 mL,混匀。
混合熔剂:无水碳酸钠和硼酸经烘干后以质量比2∶1混合,研细,并混匀。
高锰酸钾溶液:质量浓度为25 g·L-1。
草酸溶液:质量分数为1%。
硫酸锰溶液:质量分数为4%。
重铬酸钾基准试剂;五氧化二钒纯度不小于99.95%;硝酸、磷酸、硫酸、盐酸均为分析纯;试验用水为三级水。
1.2 硫酸亚铁铵标准滴定溶液的标定
准确移取3份5 mL重铬酸钾标准溶液于3个500 mL 锥形瓶中,分别依次加入50%硫酸溶液20 mL、磷酸5 mL、水70 mL,加3滴N-苯代邻氨基苯甲酸溶液,用硫酸亚铁铵标准滴定溶液滴定,溶液由紫红色变为亮绿色即为终点,不记录所消耗的体积;再加入重铬酸钾标准溶液20 mL,用硫酸亚铁铵标准滴定溶液滴定,溶液由紫红色转变为亮绿色即为终点,记录此时所消耗的硫酸亚铁铵标准滴定溶液的体积。
平行标定重铬酸钾标准溶液所消耗硫酸亚铁铵标准滴定溶液体积的极差应不超过0.10 mL,取其平均值。
硫酸亚铁铵标准滴定溶液浓度的计算公式为

式中:c为硫酸亚铁铵标准滴定溶液的浓度,mol·L-1;c1为重铬酸钾标准溶液的浓度,mol·L-1;V为所移取重铬酸钾标准溶液的体积,mL;V1为滴定重铬酸钾标准溶液所消耗的硫酸亚铁铵标准滴定溶液体积的平均值,mL。
1.3 试验原理
碳化钒样品经硝酸溶解后,加入硫酸蒸发至冒烟,冷却后加入磷酸、高锰酸钾,高锰酸钾将溶液中钒(Ⅳ)氧化成钒(Ⅴ)。用草酸除去过量的高锰酸钾,以N-苯代邻氨基苯甲酸作为指示剂,用硫酸亚铁铵标准滴定溶液进行滴定。根据硫酸亚铁铵标准滴定溶液的消耗量,计算碳化钒中钒的含量。
1.4 试验方法
1.4.1 样品分解
称取0.10 g(精确至0.0001 g)碳化钒样品于200 mL玻璃烧杯中,加入20 mL 的33%(体积分数)硝酸溶液,加热至样品溶解(有少量残渣),冷却。再用中速定量滤纸过滤,滤液收集于500 mL 烧杯中,并用热水洗涤滤纸及残渣至中性,得到的滤液作为主液保存。
接着将滤纸置于铂坩埚中,加入混合熔剂2.0 g,覆盖在其表面,放入300 ℃马弗炉中进行灰化后,于800 ℃灼烧20 min,取出冷却。用1~2滴水润湿残渣,加入50%硫酸溶液4 滴和氢氟酸2 mL,低温加热蒸发至白烟冒尽,再加17%(体积分数)盐酸溶液5 mL,继续加热浸取熔融物,将得到的浸取液合并于主液中。
1.4.2 钒的氧化及滴定
在上述所得溶液中加入50%硫酸溶液20 mL,继续加热至冒白烟2~3 min,用水稀释至100~150 mL,再加入5 mL 磷酸后,滴加高锰酸钾溶液至粉红色在2 min内不消失。接着加入硫酸锰溶液10 mL,草酸溶液0.5~1 mL,摇动至粉红色消失。加入50%硫酸溶液20 mL,并滴加2~3滴N-苯代邻氨基苯甲酸溶液,立即用硫酸亚铁铵标准滴定溶液滴定,溶液由紫红色变为亮绿色即为滴定终点,记录此时消耗的硫酸亚铁铵标准滴定溶液体积。
1.5 结果计算
碳化钒中钒的质量分数(w)为
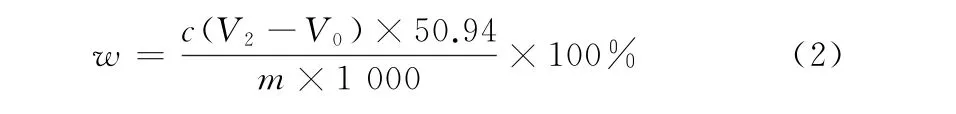
式中:c为硫酸亚铁铵标准滴定溶液的浓度,mol·L-1;V2为滴定样品时所消耗硫酸亚铁铵标准滴定溶液的体积,mL;V0为滴定样品空白时所消耗硫酸亚铁铵标准滴定溶液的体积,mL;m为样品量,g;50.94为钒的摩尔质量,g·mol-1。
2 结果与讨论
2.1 碳化钒物相分析
目前国内碳化钒制备方法主要使用冶金级五氧化二钒,与碳混合后在高温下进行反应制备。硬质合金生产中对原料杂质要求较高,本试验所使用的碳化钒采用偏钒酸铵制备[15]。图1 为碳化钒的X射线衍射(XRD)图,经jade软件分析,与标准谱图分析比对,可知碳化钒物相为V8C7。
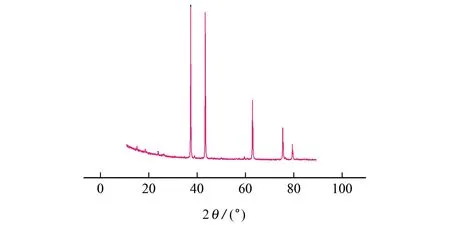
图1 碳化钒的XRD图Fig.1 XRD pattern of vanadium carbide
2.2 样品分解方法
选取碳化钒样品,分别采用酸溶解法、酸溶再碱熔残渣法(试验方法)进行分解,结果见表1。

表1 样品分解方法的对比Tab.1 Comparison of sample decomposition methods
结果表明:经硝酸溶液溶解碳化钒样品,主体已溶解,但经过滤还存在少量残渣,说明样品分解不完全,这将影响测定结果的准确度;为防止残渣中存在未溶解的碳化钒,酸溶解后再碱熔残渣,以保证碳化钒样品溶解完全。因此,试验选择酸溶再碱熔残渣法作为样品分解方法。
2.3 干扰试验
影响钒测定的元素主要有锰、铬、铈和钨[7-8]。将碳化钒样品溶解后,经ICP-AES测定[7-8]。结果表明,碳化钒中干扰元素含量较低,对主元素钒测定干扰较小,其中铝、钙、镁等质量分数均小于0.1%,锰、铬、钨和铈等干扰元素未检出。因此,碳化钒样品中钒的测定可不考虑干扰。
2.4 精密度试验
按照试验方法测定1个碳化钒样品,平行测定7次,所得钒的测定值为80.56%,计算标准偏差为0.2098%,相对标准偏差为0.26%。说明本方法测定碳化钒中钒,满足GB/T 24583.1-2019《钒氮合金 钒含量的测定 硫酸亚铁铵滴定法》的精密度要求(钒质量分数大于60.00%的重复性限为0.30%)。
2.5 方法比对
选取1 个碳化钒样品,采用国家标准方法GB/T 24583.1-2019和本试验高锰酸钾氧化-硫酸亚铁铵滴定法分别测定其中钒的含量,所得质量分数分别为80.93%,80.99%,相对误差为0.074%,结果表明本方法与GB/T 24583.1-2019测定结果基本一致。
2.6 回收试验
选用THF-1 样品为基体,加入一定量的2.0 g·L-1钒标准溶液进行加标回收试验,结果见表2。

表2 回收试验结果Tab.2 Results of test for recovery
由表2可知,钒的回收率为97.9%~100%,说明方法准确度较高。
本工作采用酸溶再碱熔残渣法处理试样后,以高锰酸钾氧化钒,用硫酸亚铁铵标准滴定溶液滴定钒,根据消耗的硫酸亚铁铵标准滴定溶液体积,最终得到碳化钒中钒的含量,方法的精密度和准确度均较高。
猜你喜欢
福建轻纺(2022年4期)2022-06-01
食品安全导刊(2021年20期)2021-11-28
化学教学(2021年6期)2021-11-17
中学生数理化(高中版.高考理化)(2020年3期)2020-05-30
环境与发展(2018年3期)2018-05-10
智富时代(2017年9期)2017-11-04
智富时代(2017年9期)2017-11-04
化学教学(2017年7期)2017-09-06
化学教学(2016年12期)2017-07-25
学周刊(2017年13期)2017-05-13
