
Regulatory role of phosphorylation in NLRP3 inflammasome activation
2021-12-04 08:54HuangYiZhouRongbin
中国科学技术大学学报 2021年5期
Huang Yi, Zhou Rongbin
1. Hefei National Laboratory for Physical Sciences at the Microscale, the CAS Key Laboratory of Innate Immunity and Chronic Disease,School of Basic Medical Sciences, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei 230027, China 2. Wuxi School of Medicine, Jiangnan University, Wuxi 214122, China
Abstract: NLRP3 is a pattern recognition receptor localized in the cytoplasm that belongs to the NOD-like receptor family. Upon activation by a wide range of danger-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs), NLRP3 recruits the adaptor protein ASC and the cysteine protease pro-caspase-1 to form a multiprotein complex called the NLRP3 inflammasome. The primary function of the NLRP3 inflammasome is to maintain homeostasis by facilitating the immune response to remove pathogens and danger signals. However, aberrant activation of the NLRP3 inflammasome also causes a variety of inflammatory diseases.Therefore, NLRP3 inflammasome activation must be precisely regulated. Recently, various kinases and phosphatases have been reported to control the NLRP3 inflammasome activation, suggesting that phosphorylation plays a vital role in regulating the inflammasome activation. In this review, we summarize how various kinases and phosphatases control the NLRP3 inflammasome activation and provide an overview of the regulatory role of phosphorylation in the NLRP3 inflammasome activation. Further, we discuss the potential pharmacologically compounds that target NLRP3-related kinases or phosphatases for the treatment of inflammasome-driven diseases.
Keywords: NLRP3 inflammasome; phosphorylation; inflammatory diseases; kinases; phosphatases
1 Introduction
NACHT, LRR and PYD domains-containing protein 3 (NLRP3) is a pattern recognition receptor (PRR) localized in the cytoplasm that belongs to the NOD-like receptor (NLR) family[1]. Upon activation by a wide range of DAMPs, such as extracellular ATP, monosodium urate (MSU), amyloid-β, silica, asbestos and dietary saturated fatty acids, and PAMPs, such as viral nucleic acids, bacterial toxins and surface proteins, NLRP3 recognizes these signals, which leads to self-oligomerization via NACHT-NACHT interactions, and the adaptor protein ASC is recruited via PYD-PYD interactions to induce the formation of a large protein speck called the ASC speck[2]. Subsequently, accumulated ASCs recruit the cysteine protease pro-caspase-1 via CARD-CARD interactions to form a multiprotein complex called the NLRP3 inflammasome[3].The NLRP3 inflammasome assembly and activation facilitate the self-cleavage and activation of pro-caspase-1. The activated caspase-1 subsequently cleaves the precursors pro-IL-1β and pro-IL-18 to produce the mature cytokines IL-1β and IL-18, respectively, and stimulate the inflammatory response. Moreover, the activated caspase-1 also cleaves Gasdermin D (GSDMD) to induce a form of cell death known as pyroptosis[4,5].Therefore, the activation of the NLRP3 inflammasome plays a crucial role in preventing the pathogen invasion and maintaining homeostasis[6]. However, overactivation of the NLRP3 inflammasome is associated with a variety of inflammatory diseases, including gout, atherosclerosis, type 2 diabetes and Alzheimer’s diseases[1]. Thus, the NLRP3 inflammasome activation must be strictly regulated.
Over the past decade,extensive efforts have been made to understand the regulatory mechanism of the NLRP3 inflammasome activation.A large number of posttranslational modifications (PTMs) have been reported to regulate the inflammasome activation, among which phosphorylation and dephosphorylation are the most extensively studied[7,8].Protein phosphorylation is a reversible process that is regulated by kinases and phosphatases, and in general,phosphorylation occurs on serine (Ser), threonine (Thr) and tyrosine (Tyr) residues via phosphoester bond formation[9,10]. In this review, we summarize how phosphorylation mediated by various kinases and phosphatases controls the activation of the NLRP3 inflammasome, including how phosphorylation regulates the activity of NLRP3 and other components of the NLRP3 inflammasome,such as ASC, pro-caspase-1 and NEK7, and provide an overview of the regulatory role of phosphorylation in the NLRP3 inflammasome activation.
2 Phosphorylation of NLRP3
The PTMs of NLRP3, which is a core component of the NLRP3 inflammasome, is essential for the assembly and activation of the inflammasome. Emerging evidence has indicated that multiple sites on NLRP3 can be phosphorylated or dephosphorylated to regulate the activation of the NLRP3 inflammasome[7,8]. Here, we summarize the phosphorylation that occurs in the PYD, NACHT, and LRR domains of NLRP3 (Figure 1).
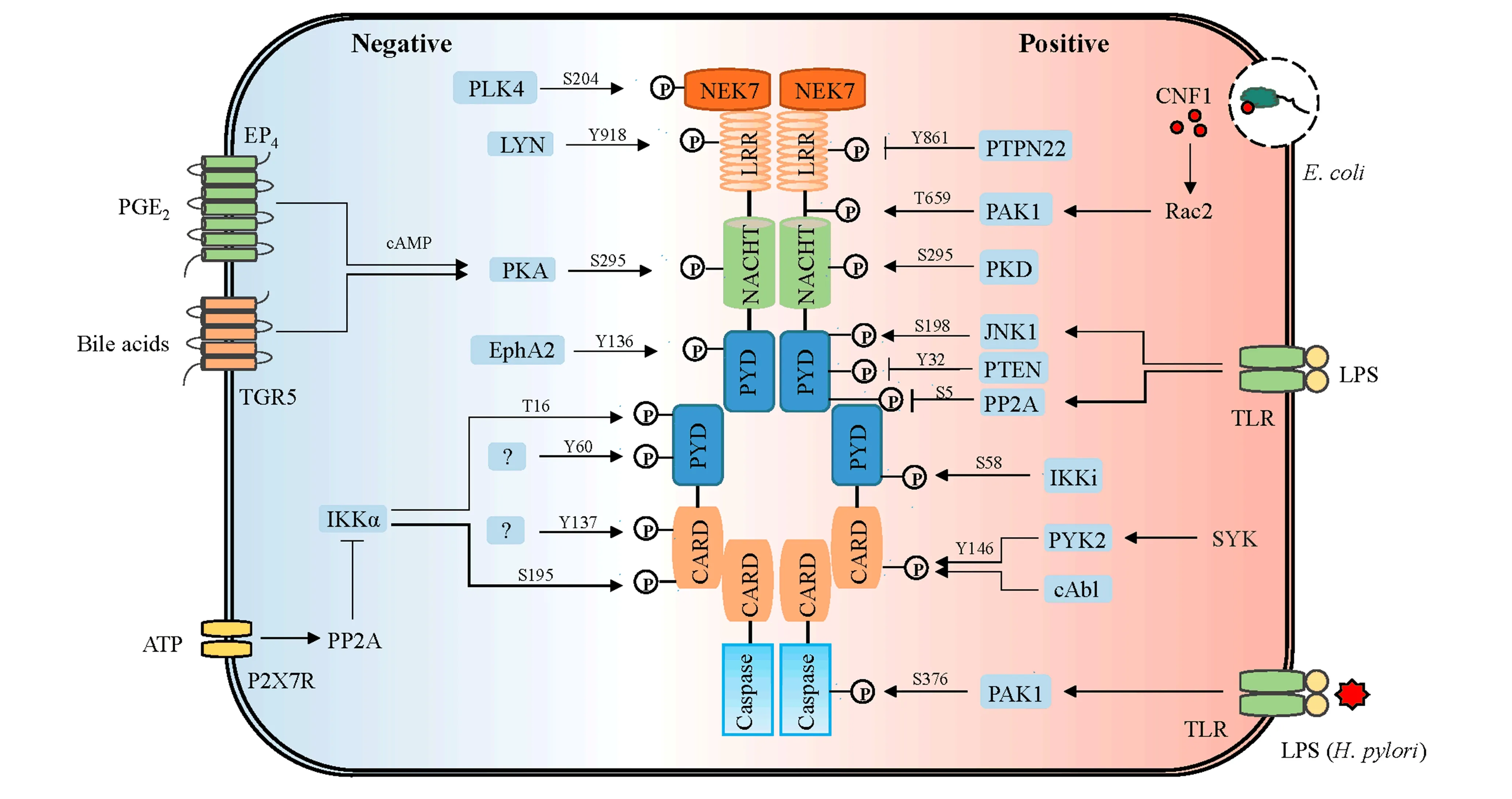
Figure 1. Regulatory role of phosphorylation in NLRP3 inflammasome activation. The phosphorylation sites of human NLRP3, ASC, Pro-caspase-1 and NEK7, and corresponding kinases and phosphatases are represented. The inhibitory effect of phosphorylation on NLRP3 inflammasome activation are indicated in black, the promoting effect of phosphorylation on NLRP3 inflammasome activation are highlighted in red, the dual effect of phosphorylation on NLRP3 inflammasome activation are shown in purple. In brief, PP2A, PTEN and PTPN22 promote inflammasome activation by dephosphorylation of NLRP3 at S5, Y32 and Y861, respectively. Lyn phosphorylates NLRP3 at Tyr918 and facilitates its ubiquitination and proteasome-mediated degradation, while Lyn can also promote the activation of NLRP3 inflammasome by inducing cathepsin B release in response toparasite plasmodium infection. NLRP3 can be phosphorylated at S295 by PKD and then released from mitochondria-associated endoplasmic reticulum membranes (MAMs), while PKA can also phosphorylates NLRP3 at S295 and inhibits NLRP3 inflammasome activation. T659 and S198 of NLRP3 can be phosphorylated by PAK1 and JNK1, respectively, promoting inflammasome activation. EphA2 binds to and phosphorylates NLRP3 at Tyr132, inhibiting ASC speck formation and inflammasome assembly. IKKα inhibites NLRP3 inflammasome activation via phosphorylating ASC at T16 and S195. Y60 and Y137 of ASC can also be phosphorylated by unknown kinases and hinding its aggregation. IKKi facilitates the translocation of ASC from the nucleus to the perinuclear via phosphorylating ASC at Ser58. Phosphorylation of ASC at Y144 by PYK2 and cAbl is critical for ASC speck formation and inflammasome activation. PAK1 phosphorylates pro-caspase-1 at Ser376 and promotes its activation. PLK4 phosphorylates NEK7 at S204 and inhibits its binding to NLRP3.
2.1 Phosphorylation of the LRR domain
The leucine-rich repeat (LRR) domain is located at the C-terminus of NLRP3, and its main function is ligand recognition and autoinhibition. Recently, the LRR domain has also been shown to be important for the interaction between NLRP3 and NEK7[11-13].Spalinger et al. reported that phosphorylation of Tyr861 (Tyr859 in mice) in the LRR domain negatively regulates the NLRP3 inflammasome activation. The researchers found that protein tyrosine phosphatase nonreceptor 22 (PTPN22) directly interacts with NLRP3 to dephosphorylate NLRP3 at Tyr861 upon the inflammasome induction by agonists and then promotes the NLRP3 inflammasome activation and subsequent IL-1β release[14]. Although NLRP3 is phosphorylated at Tyr861 after LPS priming, the kinase that directly phosphorylates NLRP3 at Tyr861 remains to be identified. In addition, it is also unclear how the NLRP3 phosphorylation at Tyr861 inhibits the inflammasome activation.Mechanistically, another study by Spalinger showed that phosphorylated but not unphosphorylated NLRP3 at Tyr861 interacts with SQSTM1 and is then recruited to phagophores.Activation of the NLRP3 inflammasome was recovered in PTPN22-deficient cells upon the loss of macroautophagy/autophagy, suggesting that phosphorylation regulates the NLRP3 inflammasome activation in an autophagy-dependent manner[15].Phosphorylation of Tyr918 (Tyr915 in mice), another important site in the LRR domain of NLRP3, also negatively regulates the inflammasome activation. Lin et al. found that the Src family protein tyrosine kinase Lyn inhibits the NLRP3 inflammasome activation by phosphorylating NLRP3 at Tyr918, which facilitates its ubiquitination and proteasome-mediated degradation[16]. Paradoxically, Shio et al. found that the tyrosine kinases Syk and Lyn play critical roles in the NLRP3 inflammasome activation induced by hemozoin(Hz), an inorganic crystal produced by the intraerythrocytic parasite Plasmodium during the heme detoxification process. Hz but not MSU-induced IL-1β production was inhibited in Lyn-deficient BMDMs pretreated with LPS[17].However, it remains unclear whether NLRP3 is directly phosphorylated by Lyn. Additionally, these two inconsistent studies suggest that the role of Lyn in the regulation of the NLRP3 inflammasome activation is complex and should be further investigated.
2.2 Phosphorylation of the NACHT domain
The nucleotide-binding-and-oligomerization (NACHT) domain is located at the center of NLRP3,and its main function is to regulate NLRP3 self-oligomerization through ATPase activities[18].Ser295 (Ser291 in mice) is an important site in the NACHT domain of NLRP3, and its phosphorylation is critical for the inflammasome activation.Mortimer et al. found that Ser295 of NLRP3 is directly phosphorylated by the kinase PKA, which is activated by prostaglandin E2 (PGE2) signaling via the PGE2 receptor E-prostanoid 4 (EP4) and inhibits the inflammasome activation by attenuating the ATPase function of NLRP3[19]. In addition, Guo et al. reported that PKA can also be activated by the bile acid signaling via the TGR5-cAMP pathway and block the inflammasome activation by promoting the NLRP3 ubiquitination after phosphorylation at Ser295[20].However, it is unknown whether the NLRP3 ATPase activity contributes to this process. Moreover, both studies suggest that PKA is a potential target for the treatment of the NLRP3 inflammasome-driven diseases.In contrast to the inhibitory effect of NLRP3 phosphorylation at Ser295 on the inflammasome activation,a recent study by Zhang et al. showed that in response to inflammasome activators, the production of diacylglycerol (DAG) in the Golgi apparatus rapidly increased as mitochondria-associated endoplasmic reticulum membranes (MAMs)localized adjacent to Golgi membranes, which recruited and activated downstream protein kinase D (PKD) to induce Ser295 phosphorylation of NLRP3, releasing self-oligomerized NLRP3 from MAMs and promoting the assembly of the active inflammasome[21].These contradictory studies suggest that NLRP3 phosphorylation at Ser295 plays dual regulatory roles in the inflammasome activation, and the mechanism remains to be further investigated. The Thr659 site (Thr657 in mice) of NLRP3 has recently been reported to be phosphorylated by p21-activated kinases 1 and 2 (Pak1/2), which is necessary for NLRP3 interactions with NEK7 and inflammasome assembly and activation[22].Further studies are needed to explore how the NLRP3 phosphorylation regulates the NLRP3-NEK7 interaction and which phosphatases mediate the dephosphorylation of NLRP3.
2.3 Phosphorylation of the PYD domain
The pyrin domain (PYD)is located at the N-terminus of NLRP3, and its main function is to recruit ASCs via the PYD-PYD interaction,which induces the inflammasome assembly[23].Stutz et al. found that NLRP3 is phosphorylated at Ser5 (Ser3 in mice) of the PYD domain in an unprimed state, which blocks charge-charge interactions between two PYDs via electrostatic repulsion and subsequent NLRP3 inflammasome assembly. Moreover, phosphatase 2A (PP2A) was found to promote the inflammasome activation by dephosphorylating the PYD domain of NLRP3[24].However, it remains unclear whether Ser5 is directly dephosphorylated by PP2A,and the kinase that phosphorylates NLRP3 at Ser5 is also unknown, thus requiring future studies. In addition to Ser5, our recent work also showed that phosphorylation at Tyr32 (Tyr30 in mice) of the PYD domain inhibited the NLRP3 inflammasome activation.The dual-specificity phosphatase PTEN promotes the inflammasome assembly and activation by directly interacting with and dephosphorylating NLRP3 at Tyr32, which enables the NLRP3-ASC interaction.The analysis of the NLRP3 mutant gene knock-in mice also showed that Tyr32 phosphorylation completely blocked the inflammasome activation[25], suggesting that phosphorylation at Tyr32 was essential for the activation of the NLRP3 inflammasome. Additionally, Zhang et al. showed that the receptor protein-tyrosine kinase EphA2 markedly inhibited the NLRP3 inflammasome activation by binding to and phosphorylating NLRP3 at Tyr132 (Tyr136 in humans) of the PYD domain, which subsequently interfered with the formation of ASC specks and the inflammasome assembly in mouse airway epithelial cells (AECs) upon the reovirus infection[26]. However, the detailed mechanism of phosphorylation at Tyr132 still requires further study.In contrast to the inhibitory effect of NLRP3 phosphorylation on the inflammasome activation, Song et al. found that phosphorylation at Ser198 (Ser194 in mice) of the PYD domain is a necessary priming event for the NLRP3 inflammasome activation.c-Jun N-terminal kinase 1 (JNK1), a downstream kinase of the TLR-IRAK1/4 pathway, promotes the inflammasome activation by mediating the phosphorylation of NLRP3 at Ser194, which is critical for NLRP3 deubiquitination[27].Recently, Ren et al. showed that the deubiquitination of NLRP3 is mediated by ABRO1, a subunit of the BRISC deubiquitinase complex, which binds to NLRP3 in an S194 phosphorylation-dependent manner and removes K63-linked ubiquitin chains from NLRP3 by recruiting the BRISC complex upon stimulation with activators[28].
Taken together, these studies suggest that multiple kinases or phosphatases regulate inflammasome activation by dynamically controlling the phosphorylation level of NLRP3. However, it is unclear how these phosphorylation modifications orchestrate the NLRP3 inflammasome activation, and the detailed mechanisms remain to be further studied.
3 Phosphorylation of other components of the NLRP3 inflammasome
In addition to NLRP3, other components of the NLRP3 inflammasome, such as ASC, caspase-1, NEK7 and DDX3X, have also been reported to be phosphorylated (dephosphorylated) by specific kinases (phosphatases) (Figure 1).
3.1 Phosphorylation of ASC
ASC is an adaptor protein that consists of an N-terminal PYD domain and a C-terminal CARD domain.Once activated, ASC binds to NLRP3 through PYD-PYD interactions and recruits pro-caspase-1 via CARD-CARD interactions, and these events essential for the assembly and activation of the NLRP3 inflammasome[2]. Recently, an increasing number of studies have shown that the phosphorylation of ASC is important for the activation and regulation of the NLRP3 inflammasome.Martin et al. found that IκB kinase α (IKKα)is a critical negative regulator of the NLRP3 inflammasome, sequestering ASC in the nucleus in resting macrophages, which requires the kinase activity of IKKα, as well as Ser193 (Ser195 in humans) and Ser16 (the same site in humans) of ASC. Upon stimulation with inflammasome activators, IKK-related kinase (IKKi) facilitates translocation of the ASC/IKKα complex from the nucleus to the perinucleus by phosphorylating ASC at Ser58 (the same site in humans). Moreover, the phosphatase PP2A is recruited to inhibit the IKKα kinase activity, allowing ASCs to dissociate from IKKα and participate in the NLRP3 inflammasome assembly and activation[29]. However, it is unclear whether IKKα directly phosphorylates ASC at Ser193 and Ser16. Another study by Mambwe et al. showed that the phosphorylation of ASC at Tyr60 (the same site in mice) and Tyr137 (no corresponding site in mice) hinders the ASC aggregation, which is an essential step for the assembly and activation of the NLRP3 inflammasome[30].However, the kinase that phosphorylates these two key sites of ASC is unknown.In contrast to the inhibitory effect of the ASC phosphorylation on the inflammasome activation,the phosphorylation of ASC at Tyr144 (Tyr146 in humans)in a Syk- and Jnk-dependent manner has been reported to be critical for speck formation and the inflammasome activation[2]. However, recent studies have shown that Syk cannot directly phosphorylate ASC. Instead, Syk phosphorylates the downstream kinase Pyk2, which is activated to directly phosphorylate ASC at Tyr144, inducing speck formation and the inflammasome activation[31]. In addition to Pyk2, Gavrilin et al. showed that the protein tyrosine kinase cAbl also promotes the inflammasome activation via the phosphorylation of ASC at Tyr144[32]. Moreover, Okada et al. found that the calcium/calmodulin-dependent protein kinase type II(CaMKII)-TAK1 signaling pathway upstream of JNK was sustained by lysosomal Ca2+release and specifically regulated the lysosome rupture-induced NLRP3 inflammasome activation[33].
3.2 Phosphorylation of pro-caspase-1
Pro-caspase--1 is acysteine protease that consists of an N-terminal caspase activation and recruitment domain (CARD) and a C-terminal caspase domain.Once stimulated by agonists,pro-caspase-1 binds to ASC via a CARD-CARD interaction and promotes the inflammasome assembly and self-cleavage and activation.The activated caspase-1 subsequently cleaves the precursors pro-IL-1β and pro-IL-18 to produce the mature cytokines IL-1β and IL-18, respectively, and boosts the inflammatory response. Moreover, the activated caspase-1 also cleaves Gasdermin D (GSDMD) to induce pyroptosis[4,5]. This finding suggests that regulating the activation of caspase-1 is crucial for the in flammasome activation.In contrast to the multiple phosphorylation sites identified in NLRP3 and ASC, only one report to date has shown caspase-1 phosphorylation. Basak et al. found that helicobacter pylori lipopolysaccharide (LPS) activated the PI-3K/Rac1/p21-activated kinase 1 (PAK1) signaling pathway in a classical TLR4-dependent manner and then induced PAK1 to directly bind to and phosphorylate caspase-1 at Ser376 (Ser374 in mice), which was crucial for caspase-1 and subsequent inflammasome activation[34].The phosphorylation of caspase-1, a key executor of the inflammasome activation, is worthy of further investigation.
3.3 Phosphorylation of NEK7
NIMA-related kinase 7 (NEK7)has been reported to act as a core component of the NLRP3 inflammasome by three separate studies[11-13], and the phosphorylation of NEK7 is also critical for the NLRP3 inflammasome activation. Yang et al. reported that polo-like kinase 4 (PLK4), the master regulator of centrosome duplication, inhibits the NLRP3 inflammasome activation by phosphorylating NEK7 at Ser204 (the same site in mice) and attenuating the interaction between NLRP3 and NEK7. Moreover, the activation of PLK4 depends on deubiquitination mediated by the deubiquitinase CYLD, which is recruited to the centrosome by the centrosomal protein Spata2[35]. This study unravels a novel link between the centrosome regulatory pathway and the NLRP3 inflammasome activation. Further studies are needed to clarify the importance of NEK7 phosphorylation in regulating the activation of the NLRP3 inflammasomes.
3.4 Phosphorylation of DDX3X
DEAD-box family member and helicase DDX3X is a newly identified component of NLRP3 inflammasome, which interacts with NLRP3 to drive the inflammasome activation and pyroptosis in response to the cellular stress[36]. It has been found that IKKε directly interacts with and phosphorylates DDX3 at ser102 (no corresponding site in mice)[37]. Moreover, IKKε facilitates the translocation of ASC from the nucleus to the perinuclear region and the NLRP3 inflammasome activation[8]. However, it is unclear whether IKKε regulates the NLRP3 inflammasome activation by inducing the DDX3X phosphorylation.
4 Potential pharmacologically compounds targeting NLRP3-related kinases or phosphatases for the treatment of inflammasome-driven diseases
The activation of the NLRP3 inflammasome has a dual function in the body. Normally, it plays a critical role in defensing against the pathogen invasion and maintaining homeostasis. However, aberrant activation of the NLRP3 inflammasome also causes a variety of inflammatory diseases, such as gout, atherosclerosis, type 2 diabetes and Alzheimer’s diseases. Therefore, the protein kinases and phosphatases that regulate the NLRP3 phosphorylation are important drug targets of those diseases. Moreover, their inhibitors may be useful in the treatment of the NLRP3 inflammasome-driven diseases.
4.1 Pharmacologically compounds targeting BTK
Bruton’s tyrosine kinase (BTK) is an essential regulator of NLRP3 inflammasome in a kinase-dependent manner, several pharmacologically drugs of BTK have been found to treat inflammasome-driven diseases by inhibiting the NLRP3 activation. The FDA-approved drug ibrutinib (PCI-32765) is a covalent inhibitor of BTK, which has been found to efficiently suppress neurological damages and infarct volume growth in a mouse model of brain ischaemia/reperfusion[38]. Mechanically, ibrutinib inhibits IL-1β release by suppressing the caspase-1 maturation and NLRP3 inflammasome activation. Moreover, ibrutinib can also inhibit the IL-1β release in human peripheral blood mononuclear cells (PBMCs) from both healthy donors and patients with CAPS as well as an S. aureus in a vivo infection model[39,40]. Consistent with this finding, Purvis et al. discovered that the ibrutinib treatment ameliorated type 2 diabetes in a high-fat food-induced mouse model by inhibiting the NLRP3 inflammasome activation[41]. In addition to ibrutinib, acalabrutinib has been found to significantly suppress the release of cytokines and chemokines, and attenuated the NLRP3 inflammasome-driven cardiac dysfunction associated with sepsis[42]. Taken together, as a well-established apharmacological target, several inhibitors of BTK are either already FDA-approved or in late-stage clinical, which are promising therapies for the inflammasome-driven diseases in the absence of the clinically approved direct inhibitors of NLRP3.
4.2 Pharmacologically compounds targeting JNK1
As an important kinase that directly regulates the NLRP3 phosphorylation, JNK has been found to be closely related to the pathogenesis of a variety of the inflammasome-driven diseases, and its targeted inhibitors are viable pharmacologically drugs for those diseases. Bentamapimod (also known as PGL5001 and AS602801) is a specific inhibitor of JNK1, which is currently in Phase II clinical trials for the treatment of the inflammatory endometriosis[43]. Although the mechanism of bentamapimod on the NLRP3 inflammasome activation is not fully understood, there is no doubt that it is an important potential pharmacologically drug for the treatment of the inflammasome-driven diseases. SP600125, another inhibitor of JNK1, was found to suppress the caspase-1 activation and cleaved-IL-1β release, and alleviate the NLRP3 inflammasome-driven peritonitis in a mouse model[33]. However,SP600125 is a broad-spectrum inhibitor of JNK, not only JNK1,but also JNK2 and JNK3. Moreover, latest studies discovered that SP600125 can also inhibit other serine/threonine kinases, including Aurora kinase A, MELK, FLT3 and TRKA, but higher concentrations are required to inhibit these kinases[44]. These results indicate that SYK inhibitors are promising for the treatment of the inflammasome-driven diseases, but more specific and safe inhibitors need to be further explored.
4.3 Pharmacologically compounds targeting other NLRP3-related kinases or phosphatases
In addition to BTK and JNK1, several other NLRP3-related kinases or phosphatases have also been found to be targets for the inflammasome-driven diseases, and their targeted compounds are promising to treatment of these diseases. It has been reported that four different inhibitors (such as CRT 0066101, Gö 6976, CID 755673, and kb NB142-70) of PKD almost completely blocked the NLRP3 inflammasome activation in peritoneal macrophages and BMDMs[21]. Moreover, those inhibitors can also suppress the NLRP3 inflammasome autoactivation in peripheral blood mononuclear cells (PBMCs) from patients with cryopyrin-associated periodic syndromes(CAPS)[21]. PF-562271,a specific inhibitor of PYK2, has been shown to dramatically abolished the NLRP3 inflammasome activation[31].Consistent with this results, PF-562271 also remission monosodium urate-mediated peritonitis in a mouse model[31].Further, two other inhibitors of PYK2, R406 and piceatannol, were also shown to inhibit the IL-1β release in BMDMs and THP-1 cells[45].
5 Concluding remarks and future perspectives
The NLRP3 inflammasome, which is composed of the innate immune receptor NLRP3, adaptor protein ASC and protease precursorpro-caspase-1, plays a crucial role in resisting the pathogen invasion and maintaining homeostasis. However, overactivation of the NLRP3 inflammasome is associated with a variety of inflammatory diseases, including gout, atherosclerosis, type 2 diabetes and Alzheimer’s diseases. Therefore, the NLRP3 inflammasome activation must be strictly regulated.Over the past decade,extensive efforts have been made to understand the mechanism by which phosphorylation regulates NLRP3 inflammasome activation.Although several kinases or phosphatases that regulate the NLRP3 inflammasome activation have been identified, the detailed mechanisms of the NLRP3 inflammasome phosphorylation remain to be further clarified. In particular, how multiple phosphorylation events orchestrate the NLRP3 inflammasome activation is unclear. Moreover, it is worth noting that there are some conflicting results regarding the role of the NLRP3 phosphorylation, which requires further investigation.Finally, understanding the regulatory mechanism of the NLRP3 inflammasome will contribute to the development of new therapies for the NLRP3 inflammasome-related diseases.

- 中国科学技术大学学报的其它文章
- A navigational system for investigating multi-sensory integration in Caenorhabditis elegans
- Carbonic anhydrase inhibitor U-104 inhibits tumor progression through CA9 and CA12 in tongue squamous cell carcinoma
- Impact of COVID-19 pandemic on stock market via sparse principal component analysis
- Pricing strategies of laborer-sharing platform in two transaction modes
- The stable subgroups of Sn acting on M0,n
- Cycle lengths in graphs of chromatic number five and six